



1. Comprensión inicial
En esta etapa, necesitamos comprender algunos conceptos y terminología, para evitar cometer errores frente a nuestros adultos mayores, como:
P: ¿Cuál es la diferencia entre RT-PCR, qPCR, PCR en tiempo real y RT-PCR en tiempo real?
Respuesta: RT-PCR es PCR de transcripción inversa(PCR de transcripción inversa, RT-PCR), que es una variante ampliamente utilizada de la reacción en cadena de la polimerasa (PCR).En RT-PCR, una hebra de ARN se transcribe inversamente en ADN complementario, que luego se usa como plantilla para la amplificación de ADN por PCR.
PCR en tiempo real y qPCR(Quantitative Real-time-PCR) son lo mismo, ambos son PCR cuantitativos en tiempo real, lo que significa que cada ciclo de PCR tiene registros de datos en tiempo real, por lo que la cantidad de plantillas iniciales se puede ajustar para un análisis preciso.
Aunque tanto la PCR en tiempo real (PCR cuantitativa fluorescente en tiempo real) como la PCR de transcripción inversa (PCR de transcripción inversa) parecen abreviarse como RT-PCR, la convención internacional es: RT-PCR se refiere específicamente a la transcripción inversaPCR, la PCR en tiempo real generalmente se abrevia como qPCR (PCR cuantitativa en tiempo real).
Y RT-PCR en tiempo real (RT-qPCR), es la PCR de transcripción inversa combinada con la tecnología cuantitativa fluorescente: primero obtenga cDNA (RT) de la transcripción inversa de RNA, y luego use PCR en tiempo real para el análisis cuantitativo (qPCR).La mayoría de los laboratorios realizan RT-qPCR, es decir, investigan sobre la regulación negativa de la expresión del ARN, por lo que la qPCR de la que todos hablan en el laboratorio en realidad se refiere a RT-qPCR, pero no olvide que todavía hay muchas pruebas de ADN en aplicaciones clínicas.Análisis cuantitativo, como la detección del VHB del virus de la hepatitis B.
Pregunta: Después de leer una gran cantidad de PCR cuantitativa fluorescente, ¿por qué el fragmento amplificado debe controlarse dentro del rango de 80-300 pb?
Respuesta: La longitud de cada secuencia de genes es diferente, algunas tienen varios kb, otras cientos de pb, pero solo necesitamos que la longitud del producto sea de 80-300 pb al diseñar cebadores, demasiado cortos o demasiado largos no son adecuados para la detección de PCR cuantitativa fluorescente.El fragmento del producto es demasiado corto para distinguirlo del dímero de cebador.La longitud del dímero de cebador es de aproximadamente 30-40 pb y es difícil distinguir si es un dímero de cebador o un producto si tiene menos de 80 pb.Si el fragmento del producto es demasiado largo, superando los 300 pb, conducirá fácilmente a una baja eficiencia de amplificación y no podrá detectar de manera efectiva la cantidad del gen.
Por ejemplo, cuando cuentas cuántas personas hay en un salón de clases, solo necesitas contar cuántas bocas hay.Lo mismo es cierto cuando detecta genes, solo necesita detectar cierta secuencia de un gen para representar. La secuencia completa servirá.Si desea contar personas, debe contar tanto la boca como la nariz, las orejas y los anteojos, y es fácil cometer errores.
Para ampliar, en la investigación biológica, hay muchos casos de investigación de un punto a otro, debido a que la secuencia de genes de cualquier especie es muy larga, es innecesario e imposible medir todos los fragmentos, como la secuenciación bacteriana 16S, que consiste en llevar a cabo la secuencia conservativa de ensayos de bacterias para inferir el número de una determinada población de bacterias.
P: ¿Cuál es la longitud óptima para el diseño de cebadores de qPCR?
Respuesta: En términos generales, la longitud del cebador es de unos 20-24 pb, lo que es mejor.Por supuesto, debemos prestar atención al valor de TM de la imprimación al diseñar la imprimación, ya que está relacionado con la temperatura óptima de recocido.Después de muchos experimentos, se ha demostrado que 60°C es un mejor valor de TM.Si la temperatura de recocido es demasiado baja, conducirá fácilmente a una amplificación no específica.Si la temperatura de recocido es demasiado alta, la eficiencia de amplificación será relativamente baja, el pico de la curva de amplificación comenzará más tarde y el valor de CT se retrasará.
P: ¿En qué se diferencia el método del tinte del método de la sonda?
Respuesta: método de tinteAlgunos colorantes fluorescentes, como SYBR Green Ⅰ, PicoGreen, BEBO, etc., no emiten luz por sí mismos, pero emiten fluorescencia después de unirse al surco menor del ADN de doble cadena.Por lo tanto, al comienzo de la reacción de PCR, la máquina no puede detectar la señal fluorescente.Cuando la reacción alcanza la etapa de hibridación-extensión, se abre la doble hebra y se sintetiza una nueva hebra bajo la acción de la ADN polimerasa, y la molécula fluorescente se une al surco menor del dsDNA.A medida que aumenta el número de ciclos de PCR, se combinan más y más colorantes con ADN de doble cadena, y la señal fluorescente también se mejora continuamente.El método del tinte se utiliza principalmente en la investigación científica.
PD: Cuidado al hacer el experimento, el tinte tiene que estar combinado con ADN humano, cuidado para convertirlo en una persona fluorescente.
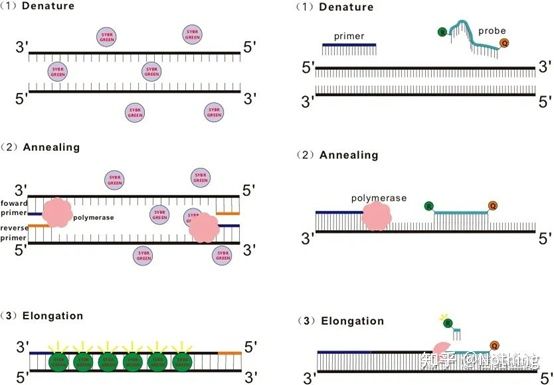
Método de colorante (izquierda) Método de sonda (derecha)
PD: Cuidado al hacer el experimento, el tinte tiene que estar combinado con ADN humano, cuidado para convertirlo en una persona fluorescente.
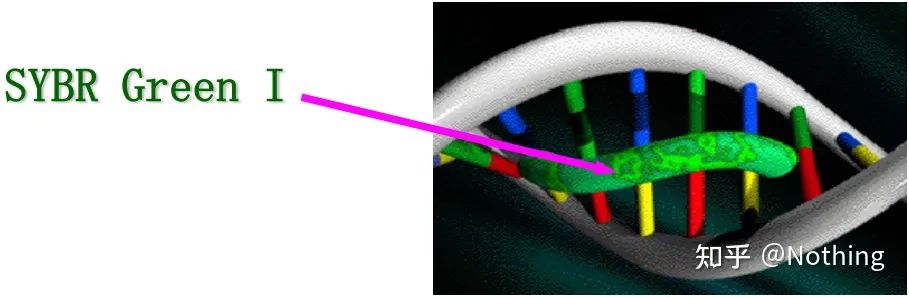
SYBR Green Ⅰ se une al surco menor del ADN
método de sondaLa sonda Taqman es la sonda de hidrólisis más utilizada.Hay un grupo fluorescente en el extremo 5 'de la sonda, generalmente FAM, y la sonda en sí es una secuencia complementaria al gen objetivo.Hay un grupo de extinción fluorescente en el extremo 3 '.De acuerdo con el principio de transferencia de energía de resonancia de fluorescencia (transferencia de energía de resonancia de Förster, FRET), cuando el grupo fluorescente informador (molécula fluorescente donante) y el grupo fluorescente de extinción (molécula fluorescente aceptora) se excitan Cuando los espectros se superponen y la distancia es muy cercana (7-10nm), la excitación de la molécula donante puede inducir la fluorescencia de la molécula aceptora, mientras que la autofluorescencia se debilita.Por lo tanto, al comienzo de la reacción de PCR, cuando la sonda está libre e intacta en el sistema, el grupo fluorescente informador no emitirá fluorescencia.Al recocer, el cebador y la sonda se unen a la plantilla.Durante la etapa de extensión, la polimerasa sintetiza continuamente nuevas cadenas.La ADN polimerasa tiene actividad de exonucleasa 5′-3′.Al llegar a la sonda, la ADN polimerasa hidrolizará la sonda de la plantilla, separará el grupo fluorescente informador del grupo fluorescente extintor y liberará la señal fluorescente.Dado que existe una relación de uno a uno entre la sonda y la plantilla, el método de la sonda es superior al método de tinción en términos de precisión y sensibilidad de la prueba.El método de la sonda se utiliza principalmente en el diagnóstico.
P: ¿Qué es la cuantificación absoluta?¿Qué es la cuantificación relativa?
Respuesta: La cuantificación absoluta se refiere al cálculo del número de copias inicial de la muestra que se analizará mediante qPCR, como cuántos virus VHB hay en 1 ml de sangre.El resultado obtenido por la cuantificación relativa es el cambio en la cantidad del gen diana en una muestra específica en relación con otra muestra de referencia, y la expresión del gen se regula hacia arriba o hacia abajo.
P: ¿La cantidad de extracción de ARN, la eficiencia de la transcripción inversa y la eficiencia de amplificación afectarán los resultados experimentales?
P: ¿El almacenamiento de muestras, los reactivos de extracción, los reactivos de transcripción inversa y los consumibles de transmisión de luz afectarán los resultados experimentales?
P: ¿Qué método puede corregir los datos experimentales?
Con respecto a estos problemas, los describiremos en detalle en las secciones avanzada y avanzada a continuación.
2. Conocimientos avanzados
Con respecto a la PCR cuantitativa fluorescente en tiempo real, debemos reconocer la realidad de que cada año se publican miles de artículos de investigación científica, entre los cuales la tecnología PCR cuantitativa fluorescente no es un número pequeño.
Si no existe un estándar común para medir el experimento de PCR cuantitativa fluorescente, los resultados pueden variar ampliamente.Para el mismo gen de la misma especie, con el mismo método de procesamiento, los resultados de detección también variarán ampliamente, y será difícil para los recién llegados repetir los mismos resultados.Tú Nadie sabe cuál está bien y cuál está mal.
¿Significa esto que la PCR cuantitativa fluorescente es una tecnología trampa o una tecnología poco fiable?No, es porque la PCR cuantitativa fluorescente es más sensible y precisa, y una pequeña operación incorrecta producirá resultados completamente opuestos.Una pequeña pérdida está a mil millas de distancia.El autor del artículo puede ser torturado repetidamente por los revisores.Al mismo tiempo, los revisores de la revista también tienen dificultades para elegir entre diferentes resultados experimentales.
En general, apunta a una falta de consenso en los experimentos de PCR en tiempo real.Con este fin, los científicos principales de la industria comenzaron a formular estándares,exigir a los contribuyentes que proporcionen algunos detalles experimentales y de procesamiento de datos necesarios (incluidos los datos necesarios) en el artículo para cumplir con estos estándares.
Los revisores pueden juzgar la calidad del experimento leyendo estos detalles;los futuros lectores también pueden usar esto para repetir el experimento o mejorarlo.Entonces los resultados experimentales obtenidos de esta manera están llenos de información, de alta calidad y utilizables.
MIBBI (Información Mínima para Investigaciones Biológicas y Biomédicas -http://www.mibbi.org) entró en vigor.MIBBI es un proyecto que proporciona estándares para experimentos.Se publica en la naturaleza.Este proyecto está dirigido a varios experimentos biológicos, incluyendo biología celular, Microarray, qPCR que vamos a discutir ahora, etc., y contempla cada tipo de experimento al enviar manuscritos.Esa información debe ser proporcionada en todo momento.
En el proyecto MIBBI, hay dos artículos relacionados con la PCR cuantitativa fluorescente, a saber:
·RDML (lenguaje de marcado de datos de PCR en tiempo real): un lenguaje estructurado y una guía de informes para datos de PCR cuantitativos en tiempo real;
·MIQE (Información mínima para la publicación de experimentos de PCR cuantitativa en tiempo real): información mínima para publicar artículos sobre experimentos de PCR cuantitativa en tiempo real.
Primero, hablemos de RDML, la especificación de terminología.
Si no hay una definición estándar para todo, es imposible continuar la discusión, por eso la explicación de los términos es tan importante en el examen.
La terminología utilizada en el experimento de PCR cuantitativa fluorescente incluye el siguiente contenido.QIAGEN nos ha hecho el mejor resumen.Los siguientes están todos secos.bienes .
Curva de amplificación
La curva de amplificación se refiere a la curva realizada durante el proceso de PCR, con el número de ciclo en abscisas y la intensidad de fluorescencia en tiempo real durante la reacción en ordenadas.
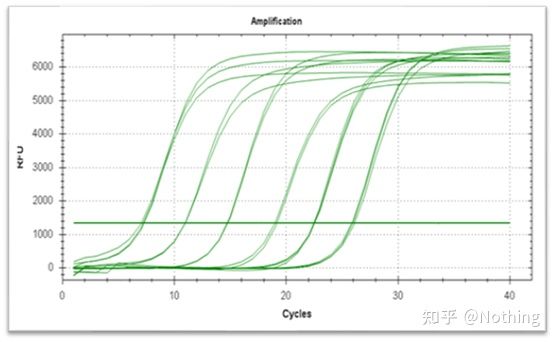
Una excelente curva de amplificación debe tener las siguientes características: la línea de base es plana o ligeramente decreciente, y no hay una tendencia alcista evidente;el punto de inflexión de la curva es claro y la pendiente de la fase exponencial es proporcional a la eficiencia de amplificación.Cuanto mayor sea la pendiente, mayor será la eficiencia de amplificación;la curva de amplificación general El paralelismo es bueno, lo que indica que la eficiencia de amplificación de cada tubo es similar;la fase exponencial de la curva de amplificación de muestras de baja concentración es obvia.
Línea de base (Línea de base)
La línea de base es el nivel de ruido del ciclo inicial, generalmente medido entre el ciclo 3 y el 15, debido a que el aumento del valor de fluorescencia causado por el producto de amplificación no se puede detectar durante este período.El número de ciclos utilizados para calcular la línea de base puede variar y es posible que deba reducirse si se utilizan cantidades altas de plantilla o si el nivel de expresión del gen objetivo es alto.
Establecer la línea de base requiere ver los datos de fluorescencia de la curva de amplificación de linealidad.La línea de base se establece de modo que el crecimiento de la curva de amplificación comience con un número de ciclo mayor que el número superior del ciclo de línea de base.Las líneas de base deben establecerse individualmente para cada secuencia objetivo.Los valores medios de fluorescencia detectados en los primeros ciclos deben restarse de los valores de fluorescencia obtenidos en los productos amplificados.Las últimas versiones de varios software de PCR en tiempo real permiten la optimización automática de la configuración de referencia para muestras individuales.
Durante los primeros ciclos de la reacción de amplificación por PCR, la señal de fluorescencia no cambia mucho.Acercarse a una línea recta se llama línea base, pero si observamos de cerca los primeros ciclos, vemos que dentro de la línea base está sucediendo lo que se muestra en la imagen a continuación.
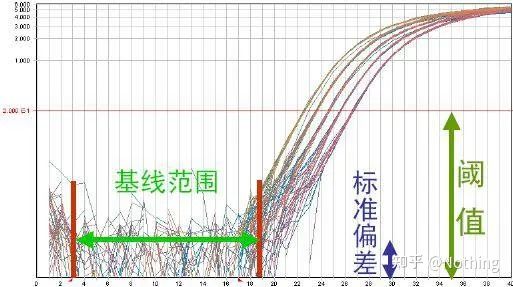
Antecedentes Antecedentes se refiere a
el valor de fluorescencia no específico en la reacción.Por ejemplo: extinción ineficaz de la fluorescencia;o una gran cantidad de plantillas de ADN de doble cadena debido al uso de SYBR Green.Los componentes de fondo de la señal se eliminan matemáticamente mediante el algoritmo del software de PCR en tiempo real.
Señal de reportero
Señal indicadora se refiere a la señal fluorescente generada por SYBR Green o sondas específicas de secuencia marcadas con fluorescencia durante la PCR en tiempo real.
Señal de reportero normalizado (RN)
RN se refiere a la intensidad de fluorescencia del colorante informador dividida por la intensidad de fluorescencia del colorante de referencia pasivo medida en cada ciclo.
Tinte de referencia pasivo
En algunas PCR en tiempo real,el colorante fluorescente ROX se utiliza como referencia interna para normalizar la señal fluorescente.Corrige las variaciones debidas al pipeteo inexacto, la posición de los pocillos y las fluctuaciones de fluorescencia pozo por pozo.

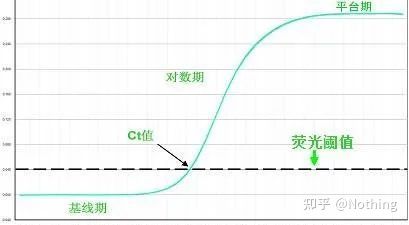
El umbral de fluorescencia (umbral)
se ajustó por encima del valor de fondo y significativamente por debajo del valor de meseta de la curva de amplificación.Debe estar en la región lineal de la curva de amplificación, que representa el rango logarítmico lineal de detección por PCR.Los umbrales deben establecerse en la vista de la curva de amplificación logarítmica para que la fase lineal logarítmica de la PCR sea fácilmente identificable.Si hay varios genes objetivo en la PCR en tiempo real, se debe establecer el umbral para cada objetivo.En general, la señal de fluorescencia de los primeros 15 ciclos de la reacción de PCR se usa como señal de fondo de fluorescencia, y el umbral de fluorescencia es 10 veces la desviación estándar de la señal de fluorescencia de los primeros 3 a 15 ciclos de PCR, y el umbral de fluorescencia se establece en la fase exponencial de la amplificación por PCR.En general, cada instrumento tiene su umbral de fluorescencia establecido antes de su uso.
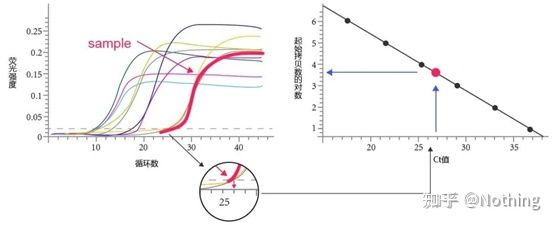
Umbral de ciclo (CT) o punto de cruce (CP)
El ciclo en el que la curva de amplificación cruza el umbral (es decir, el punto en el que la detección de fluorescencia aumenta significativamente).CT puede ser una fracción y se puede calcular la cantidad de plantilla inicial.El valor CT representa el número de ciclos experimentados cuando la señal fluorescente en cada tubo de reacción PCR alcanza el umbral establecido.Existe una relación lineal entre el valor CT de cada plantilla y el logaritmo del número de copia inicial de la plantilla, elcuanto mayor sea el número de copia inicial, menor será el valor de CT, y viceversa.Se puede hacer una curva estándar usando un estándar con un número de copia inicial conocido, donde la abscisa representa el valor de CT y la ordenada representa el logaritmo del número de copia inicial.Por lo tanto, siempre que se obtenga el valor CT de la muestra desconocida, el número de copias inicial de la muestra se puede calcular a partir de la curva estándar.
valor ΔCT
El valor de ΔCT describela diferencia entre el gen diana y el valor de CT del gen de referencia endógeno correspondiente, como un gen de limpieza, y se usa para normalizar la cantidad de plantilla utilizada:
⇒ΔCT = CT (gen diana) – CT (gen de referencia endógeno)
Valor ΔΔCT
El valor de ΔΔCT describe la diferencia entre el valor medio de ΔΔCT de una muestra de interés (p. ej., células estimuladas) y el valor medio de ΔΔCT de una muestra de referencia (p. ej., células no estimuladas).La muestra de referencia también se denomina muestra de calibración y todas las demás muestras se normalizan a esta para la cuantificación relativa:
⇒ΔΔCT = ΔCT promedio (muestra de interés) – ΔCT promedio (muestra de referencia)
Genes de referencia endógenos (genes de referencia endógenos)
Los niveles de expresión de los genes de referencia endógenos, como los genes de mantenimiento (housekeeping genes), no difieren entre las muestras.La comparación de los valores de CT del gen de referencia con el gen objetivo permite normalizar el nivel de expresión del gen objetivo a la cantidad de ARN o ADNc de entrada (consulte la sección anterior sobre los valores de ΔCT).
Genes de referencia internos correctos parala posible degradación del ARN o la presencia de inhibidores de enzimas en las muestras de ARN, así como las variaciones en el contenido de ARN, la eficiencia de la transcripción inversa, la recuperación de ácidos nucleicos y la manipulación de muestras.Para seleccionar los genes de referencia óptimos, modificamos el algoritmo para permitir su selección de la referencia óptima dependiendo del entorno experimental.
Control interno
Una secuencia de control que se amplifica en la misma reacción que la secuencia objetivo y se sonda con una sonda diferente (es decir, realizando una PCR dúplex).Los controles internos a menudo se utilizan para descartar amplificaciones fallidas, como cuando no se detecta la secuencia objetivo.
Muestra de calibración
Una muestra de referencia (por ejemplo, ARN purificado de una línea celular o tejido) utilizada en la cuantificación relativa para comparar todas las demás muestras y determinar el nivel de expresión relativo de un gen.La muestra de calibración puede ser cualquier muestra, pero normalmente es un control (por ejemplo, una muestra sin tratar o una muestra del tiempo cero del experimento).
Controles positivos
Usar reacciones de control concantidades conocidas de plantilla.Los controles positivos se utilizan a menudo para comprobar que un juego de cebadores o un juego de cebadores y sondas funcionen correctamente y que la reacción esté configurada correctamente.
Sin control de plantilla (NTC)
Una reacción de control que contiene todos los componentes necesarios de la reacción de amplificación excepto la plantilla, que generalmente se reemplaza con agua.El uso de NTC puede encontrar la contaminación causada por la contaminación del reactivo o el ADN extraño, lo que garantiza la autenticidad y confiabilidad de los datos de detección.La amplificación del control NTC indica contaminación.
Sin control RT (NRT)
El proceso de extracción de ARN puede contener ADN genómico residual, que es extremadamente dañino y es el culpable de afectar la calidad de los datos y el enemigo natural de la qPCR, por lo que al diseñar experimentos, debe diseñarse para amplificar solo la detección de ARN.Hay dos formas, una es diseñar cebadores a través de intrones, la otra es eliminar completamente el ADN, cuál es mejor, que se discutirá más adelante.El control NTR es un espejo mágico para detectar la contaminación del ADN.Si hay amplificación, significa que hay contaminación.
Estándares
Los estándares son muestras de concentración conocida o número de copias que se utilizan para construir una curva estándar.Para garantizar la estabilidad del estándar, el fragmento del gen generalmente se clona en el plásmido y se usa como estándar.
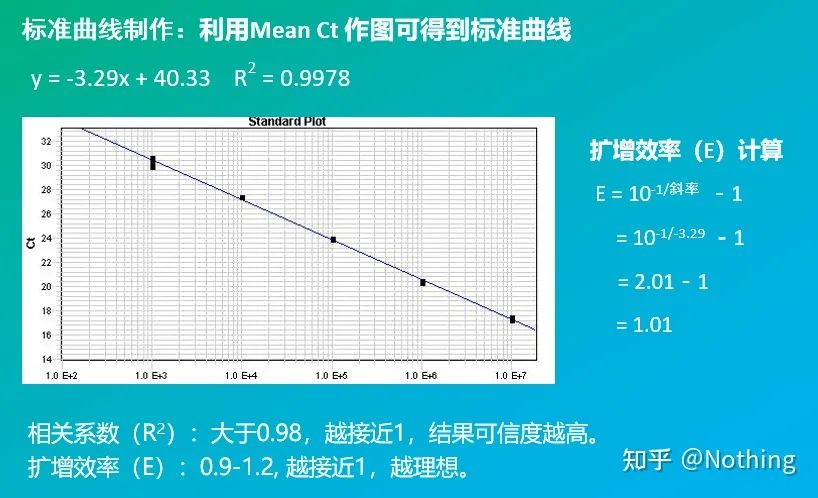
La curva estándar
generalmente se diluye en al menos 5 gradientes de concentración con el producto estándar de acuerdo con la proporción de duplicación, y se dibujan 5 puntos en las coordenadas del valor de CT y el número de copias, y los puntos se conectan para formar una línea para generar una curva estándar.Para cada curva estándar, se debe verificar su validez.El valor de la pendiente cae entre -3,3 y -3,8, y cada concentración se realiza por triplicado.Los puntos que sean significativamente diferentes de otros puntos deben descartarse.El valor de CT de la muestra que se analizará se lleva a la curva estándar y se puede calcular el nivel de expresión de la muestra que se analizará.
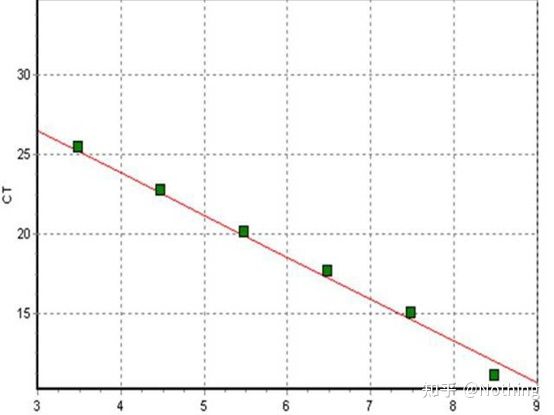
El valor CT de la muestra que se va a analizar se introduce en la curva estándar y se puede calcular el número de copias inicial de la muestra que se va a analizar.
Eficiencia y Pendiente
La pendiente de la curva estándar representa la eficiencia de la PCR en tiempo real.
·Una pendiente de -3.322 indica que la eficiencia de amplificación de PCR es 1, o 100% eficiente, y la cantidad de producto de PCR se duplica en cada ciclo.
·Una pendiente inferior a -3,322 (p. ej., -3,8) indica una eficiencia de PCR
·Una pendiente mayor a -3.322 (por ejemplo, -3.0) indica que la eficiencia de la PCR parece ser mayor al 100%, lo cual es curioso, ¿cómo un ciclo de PCR podría generar más del doble del producto amplificado?Esta situación se da en la fase no lineal de la reacción de PCR, es decir, hay una gran cantidad de amplificación no específica.
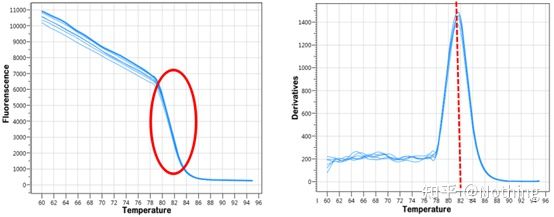
curva de fusión
Una vez completada la amplificación de qPCR, el producto de PCR se calienta.A medida que aumenta la temperatura, el producto de amplificación de doble cadena se derrite gradualmente, lo que provoca una disminución de la intensidad de la fluorescencia.Cuando se alcanza una cierta temperatura (Tm), una gran cantidad de productos se derretirán.La fluorescencia cae bruscamente.Diferentes productos de PCR tienen diferentes valores de Tm y diferentes temperaturas de fusión, por lo que se puede identificar la especificidad de la PCR.
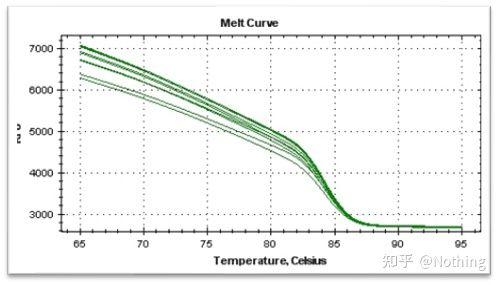
Curva de fusión (curva derivada)
La curva de fusión se deriva para formar un mapa de picos, que puede mostrar de manera más intuitiva la situación de los fragmentos de productos de PCR.Dado que la temperatura de fusión es el valor de Tm del fragmento de ADN, se pueden juzgar algunos parámetros que afectan el valor de Tm del fragmento de ADN, como el tamaño del fragmento, el contenido de GC, etc. En términos generales, de acuerdo con nuestros principios de diseño de cebadores,la longitud del producto amplificado está en el rango de 80-300bp, por lo que la temperatura de fusión debe estar entre 80°C y 90°C.
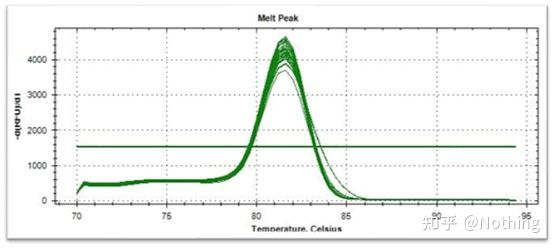
Interpretación de la curva de fusión: Si el único pico principal aparece entre 80°C-90°C, significa que la PCR cuantitativa fluorescente es perfecta;si el pico principal aparece entre 80 °C y 90 °C y varios picos aparecen por debajo de 80 °C, se considera básicamente el dímero del cebador.Puedes intentar aumentar la temperatura de recocido para solucionarlo;si el pico principal aparece entre 80°C y 90°C, y el pico misceláneo vuelve a aparecer cuando la temperatura aumenta, básicamente se considera que hay contaminación de ADN, y el ADN debe eliminarse en la etapa inicial del experimento.
Por supuesto, todavía hay algunas situaciones anormales, que se desglosarán una por una a continuación.
3. Conocimientos avanzados
Para hacer qPCR, tengo que decir MIQE,Información mínimapara Publicación deCuantitativoPCR en tiempo realExperimentos: la información mínima para publicar artículos sobre PCR cuantitativa en tiempo realexperimentosPara simplificar la comprensión de todos, simplificaremos el contenido clave.
El texto original del MIQE lo puedes buscar en Internet, y lo más importante es que estipula ellista de verificación de datos que debe proporcionarse al publicar un artículo .
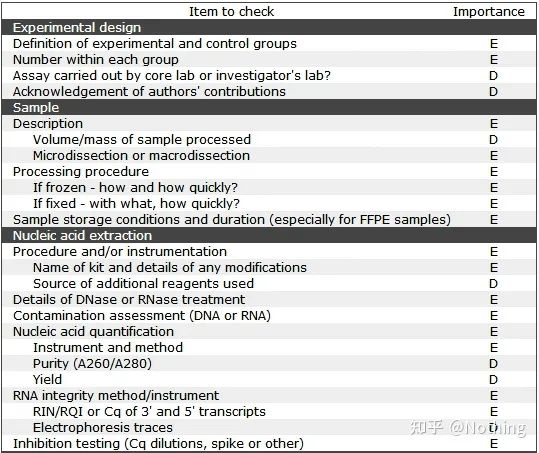
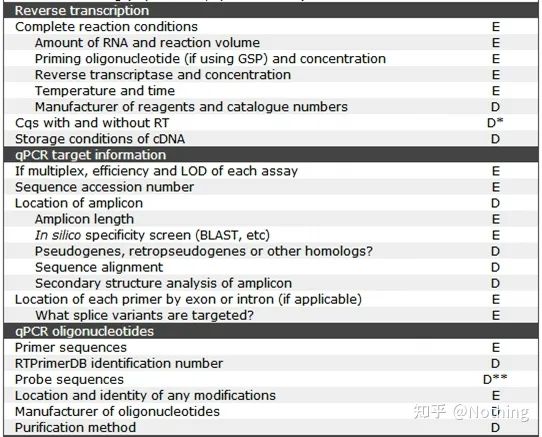
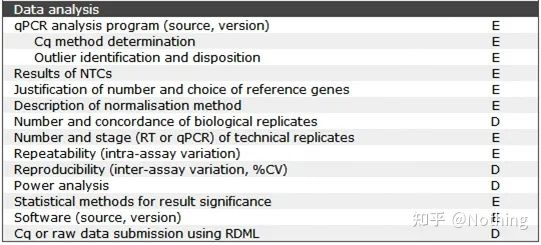
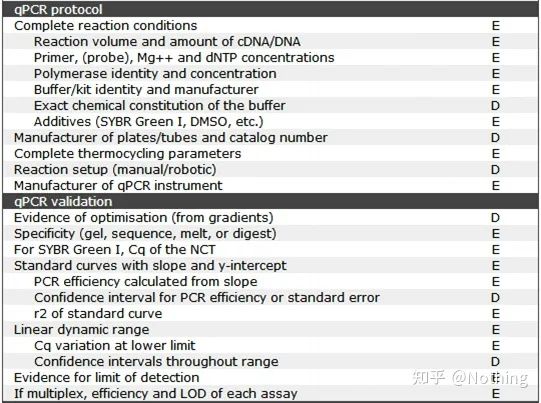
Los revisores pueden juzgar la calidad del experimento leyendo estos detalles;los futuros lectores también pueden usar esto para repetir o mejorar el experimento.
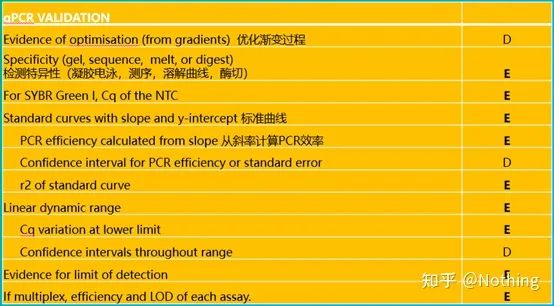
Vale la pena señalar que en esta lista, la importancia de cada lista está marcada con E o D respectivamente.¿Qué significa?E: información esencial (debe ser enviada);D: información deseable (proporcionar la mayor cantidad posible).
MIQE (1)—Diseño Experimental
Muchos cabrones que han completado su defensa después de terminar sus estudios de posgrado no sabrán cómo diseñar un experimento de forma independiente, abrir sus cuadernos y hacer lo que el maestro les dice que hagan.Como resultado, el diseño experimental no fue riguroso, y el departamento editorial de la revista dijo que querían inventar esta foto y esa foto, así que lo hicieron aturdidos.¡Así se hacen los cabrones!

Más cerca de casa, el primer principio del experimento es determinarel rigor de la lógica experimental.Lo más fundamental es el diseño experimental, y lo más importante del diseño experimental es cómo establecer la muestra objetivo, la muestra de referencia (control) y el número de repeticiones, de modo que los datos experimentales puedan ser referenciados, comparables y convincentes.
La muestra objetivose refiere a la muestra que nos obliga a detectar el gen diana tras un determinado tratamiento.La muestra de referenciaes la muestra sin ningún tratamiento, que a menudo se denomina tipo salvaje en biología.
Repeticiones experimentalesEs muy importante.Generalmente, el número de réplicas persuasivas debe ser más de tres.Es necesario distinguir qué es la replicación biológica y qué es la replicación técnica.
Réplicas biológicas: El mismo experimento de verificación realizado con diferentes materiales (tiempo, plantas, lotes, placas de reacción).

Duplicación biológica
Tomemos como ejemplo el tratamiento con pesticidas de la pimienta.Queremos rociar pesticidas en las tres plantas de ABC, entonces las tres plantas de ABC son tres réplicas biológicas, y son el mismo experimento de verificación realizado con diferentes materiales.Pero como experimento, definitivamente se necesita un control, por lo que podemos rociar una de las ramas de la planta A para formar un grupo experimental de la planta A, y no rociar las otras ramas de la planta A para formar un grupo de control.Haz lo mismo para B y C.
Réplicas Técnicas (Réplicas Técnicas): Es un experimento repetido diseñado para evitar errores causados por la operación, que en realidad es un agujero duplicado incluido en el mismo material.Tanto los tratamientos como los controles deben tener configuraciones replicadas (mínimo tres) del gen objetivo y el gen de referencia interno.
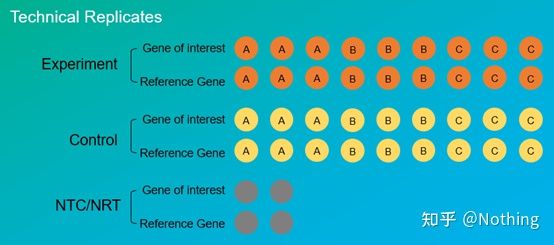
Repetición técnica
Tome la pimienta tratada con pesticidas como ejemplo nuevamente.Para el grupo experimental de la planta A, hicimos tres hoyos de PCR de 1, 2 y 3 para su gen objetivo y su gen de referencia interno respectivamente, para tomar el promedio después de la detección.Para el control de la planta A, los Grupos también se tratan de la misma manera.Del mismo modo, haga el mismo tratamiento para las plantas B y C.Esto es repetición técnica.
Cabe resaltar quelo que entra en la estadística es la repetición biológica, y la repetición técnica es para probar si hay algún fenómeno aleatorio en el proceso experimental, para hacer creíbles los resultados experimentales, es decir, para evitar errores tomando su promedio como solemos decir.
Controles negativos—NTC y NRT
NTC (control sin plantilla), un control sin plantilla, se utiliza para verificar si el material experimental está contaminado.Generalmente, el agua se utiliza como plantilla.Si hay una reacción fluorescente, indica que se ha producido una contaminación de ácido nucleico en el laboratorio.
Estas contaminaciones provienen de: agua impura, reactivos no calificados que contienen ADN endógeno, contaminación por cebadores, contaminación por equipos de laboratorio, contaminación por aerosoles, etc., es necesario utilizar eliminadores de RNasa e inhibidores de RNasa.La contaminación por aerosoles es la más difícil de encontrar.Imagina que tu laboratorio es como el smog, con varios ácidos nucleicos suspendidos en el aire.

NRT (sin transcriptasa inversa), el control sin transcripción inversa, es el ARN con transcripción no inversa como control negativo, que es el control del residuo de ADNg.
Al realizar la expresión génica, la cantidad de ARN se detecta detectando la cantidad de ADNc después de la transcripción inversa.Si hay residuos de gDNA cuando se purifica el RNA, se producirán errores en los resultados experimentales, porque los resultados reales obtenidos son gDNA y cDNA.A nivel agregado, no solo el ADNc, el ADNg debe eliminarse por completo durante la extracción del ARN.
MIQE (2): información de muestra
La llamada información de muestra significa que cuando publicamos un artículo sobre qPCR, debemos explicar claramente la información de muestra, que es una parte indispensable del artículo.De manera similar, cuando procesamos muestras, también debemos regular nuestras propias operaciones para garantizar la validez de las muestras.

La descripción de la muestra es solo un resultado, y debemos prestar más atención a los materiales tomados durante todo el experimento.
Selección de materiales experimentales
Muestras de sangre: elija sangre fresca, no más de 4 horas.Muestras de células: elija recolectar células frescas en un período de crecimiento vigoroso.Tejido animal: elija tejido fresco que crezca vigorosamente.Tejido vegetal: elija tejido fresco y joven.
Debes haber notado que hay una palabra clave en estas pocas oraciones: fresco.
Para las muestras anteriores, el mejor kit, rentable y estable del mercado es el kit de Foregene, que puede extraer rápida y fácilmente su ADN y ARN.
Kit de aislamiento de ARN total de células
Kit de aislamiento de ARN total animal
Kit de aislamiento de ARN total de plantas
Kit de aislamiento de ARN total de plantas Plus
Kit de aislamiento de ADN vegetal
Almacenamiento de materiales experimentales
En términos generales, no recomendamos almacenar muestras, si las condiciones lo permiten.Sin embargo, hay muchos amigos que no pueden realizar experimentos inmediatamente después del muestreo, y algunos incluso necesitan llevar tanques de nitrógeno líquido al campo para realizar el muestreo.
Para este tipo de amigo trabajador, solo puedo decir que no entiende los consumibles de reactivos.Ahora, muchas empresas de consumibles de reactivos producen reactivos que pueden almacenar muestras de ARN a temperatura ambiente y usted puede optar por utilizarlos.El método de almacenamiento convencional es el almacenamiento de nitrógeno líquido, utilizando un pequeño tanque de nitrógeno líquido que es fácil de transportar.Después de llevar la muestra al laboratorio, guárdela en un refrigerador a -80°C.

Para experimentos que involucren ARN, se debe seguir el principio de seis palabras:baja temperatura, sin enzimas,yrápido .
El concepto de baja temperatura es fácil de entender;sin enzimas, la RNasa está en todas partes del mundo en el que vivimos (de lo contrario, el VIH lo habría matado), por lo tanto, cómo evitar la RNasa al hacer experimentos es un concepto muy importante;rápido,No hay Kung Fu en el mundo que no se pueda romper, solo la velocidad no se puede romper..
Por lo tanto, en cierto sentido, cuanto más corto sea el tiempo de extracción, mejor será el kit.Por queForegeneLa equipación enfatiza la velocidad, porque la conocen bien.
PD: Algunas chicas hacen experimentos con mucho cuidado, pero no son tan buenos como una volcada después de varios años de trabajo.Sienten que Dios es injusto, quejándose de los demás y buscando la vida.De hecho, ella no lo entendía.No protegió bien al ARN, y el jugador de volcadas era ágil.Cuando estaba haciendo el experimento pensó que terminaría el slam dunk con tres tiempos, cinco tiempos y dos divisiones, pero hizo bien el experimento.
Nota: Más lento, más posibilidades de invasión de RNase.¿Cómo entrenarte para ser rápido?No hay manera, solo practica más.
Para diferentes experimentos y diferentes muestras, todavía es necesario leer más literatura y elegir un método apropiado para el procesamiento.Para el proceso de recolección y almacenamiento de muestras, MIQE requiere que se escriba claramente en el papel, para que los revisores puedan revisar la confiabilidad del papel, y también es conveniente que los jóvenes atónitos repitan su experimento.
Aunque los experimentos biológicos son difíciles, son de alta gama.Si no tienes cuidado, puedes volcar el mundo.Por ejemplo, convertir el SARS en una crisis bioquímica o hacer arroz híbrido para salvar a 1.300 millones de personas.La imagen de abajo es un experimento químico, debes entender lo orgulloso que estás de tu investigación con solo mirar su apariencia de pene.Olvídalo, no lo ennegrezcas.

MIQE (3) – extracción de ácidos nucleicos.
La extracción de ácidos nucleicos es un gran evento y todos los experimentos de biología molecular comienzan con la extracción de ácidos nucleicos.En primer lugar, copiemos el contenido de MIQE sobre la extracción de ácidos nucleicos.
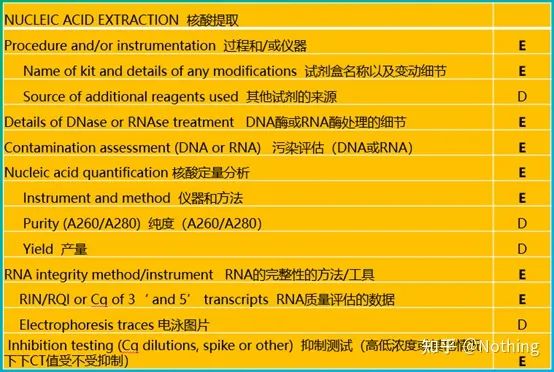
Mirando esta forma, no puedes quedarte en la superficie.La forma es un dogma.Para ser un estudiante destacado, debe preguntarse por qué.El contenido esencial de esta tabla es: Perseguirla pureza, integridad, consistencia y cantidad de extracción de ARN .
La primera parte de laproceso o instrumento es el paso de extracción de ácido nucleico.Si usa un extractor automático de ácido nucleico para extraer (avanzado, contácteme para la compra), debe indicar el nombre del modelo del instrumento.

El nombre del equipo y
Los detalles de qué kit se utilizó para el cambio, qué reactivos especiales se agregaron o qué operaciones especiales se realizaron deben explicarse claramente para que otros puedan repetir fácilmente su experimento.
Algunas personas agregan algunos reactivos especiales cuando extraen muestras especiales, pensando que esta es su arma secreta y no se lo dicen a los demás.Mientras lo mantienen en secreto, también pierden la oportunidad de hacer brillar su artículo.No seas inteligente, tienes que ser más honesto que el viejo Zhang en la investigación científica, si quieres ser inteligente, el artículo te hará estúpido.
debe recordar el número de producto del kitcuando pides el kit y escribes el artículo.En general, hay dos números en el kit: Cat: número de catálogo (número de producto, número de artículo), Lot: número de lote del producto (se usa para indicar de qué lote proviene el producto).
Además, el número CAS se usa a menudo al pedir reactivos bioquímicos, y lo popularizaré juntos.El número CAS es el número asignado por la American Chemical Society a cada nuevo fármaco químico.Generalmente, tres números están conectados por un guión.Número CAS de Rushui: 7732-18-5.Los productos químicos suelen tener múltiples alias, pero el número CAS es único.Al pedir un medicamento, primero puede verificar su número CAS.
Más cerca de casa, ¿por qué tenemos que describir estas cosas claramente?De hecho, también es para comprobar la calidad de la extracción de ARN.El uso de instrumentos y kits hará que la extracción de ARN sea más consistente.La escala de extracción de los laboratorios ordinarios no es grande y se puede obtener con kits.
Los detalles del tratamiento con ADNasa o ARNasa
La cuestión importante de la PCR cuantitativa fluorescente es evitar la contaminación del ADN y no experimentar si hay contaminación.Por lo tanto, es imperativo indicar el proceso que usó para procesar el ADN, a fin de demostrar que el ADN en el proceso experimental se eliminó total y completamente.representada por un diagrama esquemático.
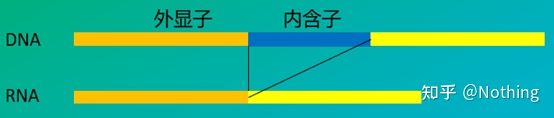
Diagrama esquemático de ARN y ADN.
En general, el método para eliminar el ADN es tratar el ARN con ADNasa después de la extracción.Sin embargo, estos son métodos relativamente antiguos.Los kits comerciales de extracción de ARN han podido eliminar el ADN durante el proceso de extracción sin agregar ADNasa.Por ejemplo, una serie de kits de Foregene.
Nota: Eliminar el ADN durante la extracción del ARN es un arma de doble filo muy peligrosa, que prolongará el tiempo de operación de la extracción del ARN y aumentará el riesgo de degradación del ARN.Básicamente, es una compensación entre el rendimiento y la pureza del ARN.
Además, la cantidad de ADNasa añadida a la columna de adsorción basada en sílice es muy pequeña y se debe utilizar ADNasa de alta calidad para lograr el efecto.La ADNasa no optimizada no se puede digerir rápida y completamente.Esta es una prueba del nivel técnico del comerciante.Por supuesto, hay comerciantes aún más extraños que se jactan de que el ADN se puede eliminar sin ADNasa.Se puede decir que cualquiera que se jacte de que el ADN se puede eliminar por completo sin ADNasa es un gamberro.El ADN es una estructura de doble cadena relativamente estable y no se puede eliminar simplemente hablando y riendo.
Evaluación de la contaminación
método de evaluación: detección por electroforesis, agarosa al 1 %, 6 V/cm, 15 min, carga 1-3 ul
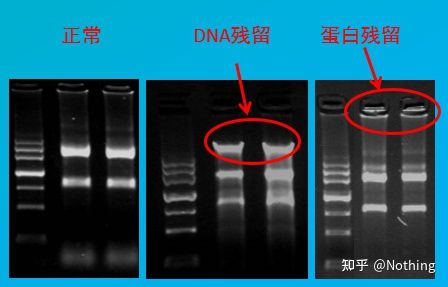
Análisis cuantitativo de ácidos nucleicos
generalmente se mide usando un espectrofotómetro UV.Permítanme primero popularizar el significado de los tres valores de OD260, OD280 y OD230.
·OD260nm: Es la longitud de onda de absorción del pico de absorción más alto de ácido nucleico, y el mejor valor medido oscila entre 0,1 y 1,0.De lo contrario, diluya o concentre la muestra para que esté dentro del rango.
·OD280nm: Es la longitud de onda de absorción del pico de absorción más alto de proteínas y sustancias fenólicas.
·OD230nm: Es la longitud de onda de absorción del pico de mayor absorción de carbohidratos.
A continuación, hablemos del papel de cada indicador.Para A260, se puede utilizar para medir el rendimiento de ácido nucleico.Cuando OD260 = 1, dsDNA = 50 μg/ml, ssDNA = 37 μg/ml, RNA = 40 μg/ml.
Para la pureza, debemos observar las proporciones que comúnmente vemos: OD260/280 y OD260/230.
·ADN puro: OD260/280 es aproximadamente igual a 1,8.Cuando es mayor a 1,9 indica que hay contaminación por ARN, y cuando es menor a 1,6 indica que hay contaminación por proteínas y fenoles.
·ARN puro: 1,7
·OD260/230: Ya sea ADN o ARN, el valor de referencia es 2,5.Cuando es menor a 2.0 indica que hay contaminación de azúcar, sal y materia orgánica.
integridad del ARN
Es muy importante medir la integridad del ARN.Generalmente, es necesario hacer un experimento de gel de desnaturalización de ARN para verificar si el brillo entre el ARN 28S y 18S es una relación doble.Cuando aparece la tercera banda 5S, significa que el ARN ha comenzado a degradarse, excepto en los invertebrados.

Datos para la evaluación de la calidad del ARN: además de las pruebas anteriores, también hay algunas pruebas de instrumentos más avanzadas en términos de integridad del ARN, como la prueba de integridad RQI del sistema de electroforesis automática Experion, que puede detectar si el ARN se degrada de forma invisible.
En la investigación científica, la PCR cuantitativa fluorescente es una comparación entre el gen objetivo y el gen de referencia interno.Por lo tanto, en el proceso de conservación de muestras de ARN, extracción de ARN, etc., el objetivo principal es garantizar la integridad del ARN.
La forma en que la integridad del ARN afecta el equilibrio entre el gen objetivo y el gen de referencia interno se puede entender fácilmente a partir de la figura a continuación.La degradación conducirá a la incompletitud del gen, ya sea la incompletitud del gen de referencia interno o la incompletitud del gen objetivo, tendrá un gran impacto en los datos.

El diagrama esquemático del gen objetivo y el gen de referencia no debe ser verdadero
Prueba de inhibición (si el valor de CT se suprime en condiciones de alta o baja concentración u otras condiciones)
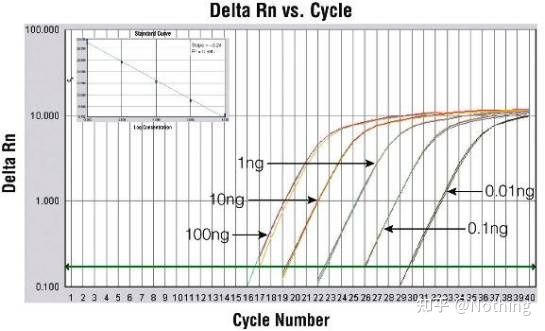
Tomando esta figura como ejemplo, los valores de Ct de las cinco curvas son los siguientes.La distribución de los valores de CT entre las curvas es desigual, y los valores de Ct se retrasan en concentraciones altas y bajas, que es el caso de la inhibición de la PCR.
Punto clave: en el proceso de extracción de ARN, debemos abandonar los conceptos erróneos y establecer los correctos.
La idea equivocada es: la extracción de ARN solo persigue el rendimiento, pensando que cuanto mayor sea la cantidad de ARN obtenido, mejor.De hecho, cuando hacemos la cuantificación, si el número de genes no es muy grande, no necesitamos mucho ARN.La cantidad de ARN que extraes es más que suficiente.
El concepto correcto es:La extracción de ARN debe perseguir la pureza, la integridad y la consistencia..La pureza puede garantizar que la transcripción inversa posterior no se inhiba y que los datos no se vean afectados por el ADN.La integridad garantiza el equilibrio de las secuencias objetivo y las referencias internas.La consistencia asegura una carga de muestra estable.
MIQE (4) – transcripción inversa
Idea equivocada: la búsqueda de un mayor volumen de muestra.
concepto correcto: Persiga la consistencia (estabilidad), independientemente de la cantidad de ARN cargado, la eficiencia de la transcripción inversa se mantiene constante, lo que garantiza que las diferencias en el ADNc realmente reflejen las diferencias en el ARNm.
Explicamos este proceso con un diagrama esquemático:
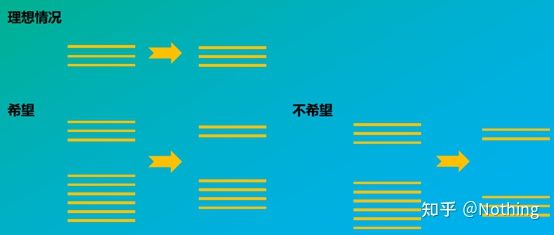
Diagrama esquemático de la eficiencia de la transcripción inversa, no sea cierto
En primer lugar, debemos comprender la diferencia entre el proceso de transcripción inversa y el proceso de PCR.La PCR se somete a múltiples procesos de calentamiento y recocido, y el fragmento objetivo crece exponencialmente;mientras que la transcripción inversa no tiene este proceso, podemos imaginar que la transcripción inversa es en realidad uno a uno Durante el proceso de replicación, tantas piezas de ARN
Como se pueden obtener tantas piezas de información de cDNA, debería entenderse ahora, porque los fragmentos grandes y pequeños se han transcrito inversamente y es imposible concentrarse en un fragmento.Y debido a que la cantidad de ARN es relativamente pequeña, la cantidad de ADNc obtenido también es relativamente pequeña, a diferencia de la PCR, que tiene un efecto de amplificación, por lo que es básicamente imposible de detectar.
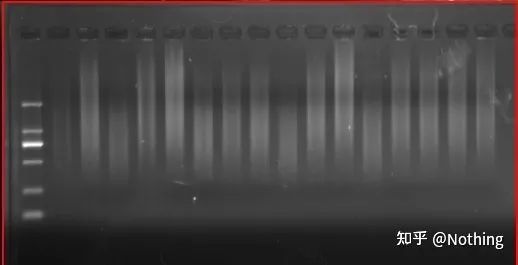
resultados de la electroforesis de ADNc
En segundo lugar, idealmente, la transcripción inversa se realiza uno a uno, pero ninguna transcriptasa inversa de ninguna compañía puede lograr este efecto.Básicamente, la eficiencia de la mayoría de las transcriptasas inversas oscila entre el 30 y el 50 %.Si este es el caso, preferiríamos tener una eficiencia de transcripción inversa relativamente estable, que es lo que queremos ver en la figura: 3 ARN obtienen 2 ADNc, 6 ARN obtienen 4 ADNc, por lo que no importa cuánta muestra se cargue, la eficiencia de transcripción inversa es relativamente estable.No queremos ver la situación en la que la eficiencia de la transcripción inversa es inestable y se inhibe la alta concentración.
Entonces, ¿cómo verificar si la eficiencia de la transcripción inversa es estable?El método es muy simple, solo necesita hacer una prueba de comparación: una es transcribir inversamente en cDNA después de duplicar la dilución de RNA, y la otra es duplicar la dilución después de transcribir inversamente en cDNA, y luego hacer qPCR para ver si la pendiente obtenida es consistente.Como estudiante superior, deberías entenderlo en segundos.Como se muestra abajo:
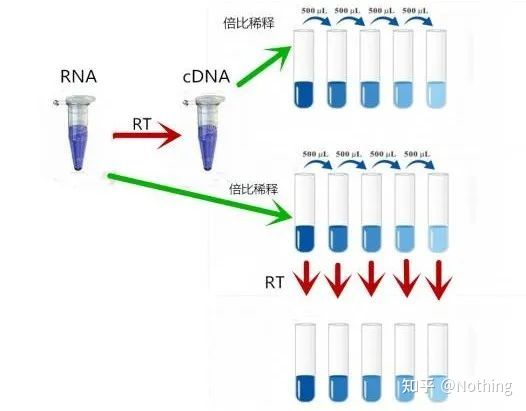
Dilución de ARN y ADNc para probar si la eficiencia de la transcripción inversa es estable
transcriptasa inversa y kit
¿Cómo puede la PCR cuantitativa fluorescente perfecta tener una excelente transcriptasa inversa y un kit?La transcriptasa inversa se divide aproximadamente en dos tipos según la fuente, AMV oM-MLV, y su rendimiento es el mismo que el que se muestra en la tabla.
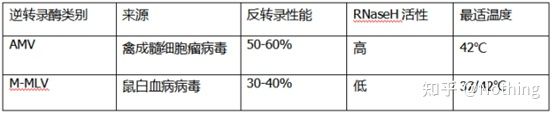
Actividad de ARNasa H
La RNasa H es Ribonucleasa H, el nombre chino es ribonucleasa H, que es una endorribonucleasa que puede hidrolizar específicamente el ARN en la cadena híbrida ADN-ARN.La RNasa H no puede hidrolizar los enlaces fosfodiéster en el ADN o ARN monocatenario o bicatenario, es decir, no puede digerir el ADN o ARN monocatenario o bicatenario.Comúnmente utilizado en la síntesis de la segunda cadena de cDNA.
Es una cosa extraña.Decimos que la transcriptasa inversa tiene actividad de RNasa H, no que la transcriptasa inversa contiene RNasa H, y puede que no sea posible separar la RNasa H de la transcriptasa inversa, quizás debido a la conformación de ciertos grupos en la transcriptasa inversa. Esta actividad es causada por la transcriptasa inversa.
Por lo tanto, independientemente de la mayor eficiencia de transcripción inversa de AMV, su actividad RNase H reduce el rendimiento de cDNA.Por supuesto, los fabricantes de reactivos optimizan constantemente sus productos para eliminar la actividad de la RNasa H en la transcriptasa inversa tanto como sea posible para aumentar el rendimiento del ADNc.
Temperatura de recocido
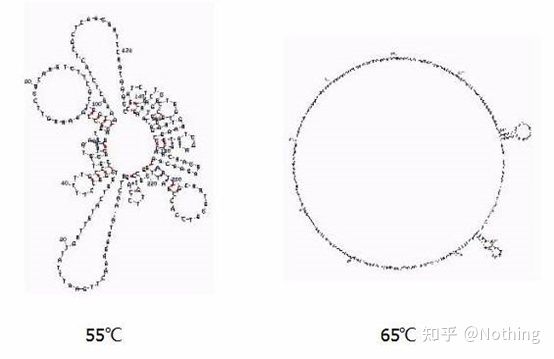
Estructura secundaria del ARN a diferentes temperaturas.
Consulte la figura anterior para ver la estructura secundaria del ARN a diferentes temperaturas y use la herramienta en línea mFold para determinar la estructura secundaria del fragmento objetivo en condiciones específicas de temperatura y concentración de sal.A 55 °C, la estructura secundaria del ARN sigue siendo muy compleja, la transcriptasa inversa no puede funcionar y la estructura secundaria no puede resolverse por completo hasta los 65 °C, mientras que la temperatura óptima de AMV y M-MLV es mucho más baja que esta temperatura.
¿qué hacer?La estructura secundaria es el apareamiento complementario de la propia plantilla, lo que conduce a una fuerte competencia entre el cebador y la transcriptasa inversa y la plantilla, lo que da como resultado una serie de problemas como baja E y mala repetibilidad.
¿qué hacer?Solo aumente la temperatura de recocido tanto como sea posible.
Muchos fabricantes de reactivos están mejorando su transcriptasa inversa a través de la ingeniería genética.Algunos aumentan la temperatura de reacción, como Jifan y Aidelai, y algunos eliminan el grupo activo de la enzima RNasa H para mejorar la afinidad entre la enzima y la plantilla de ARN.La alta afinidad puede exprimir competitivamente la estructura secundaria y leer sin problemas, y también mejorar en gran medida la eficiencia de la transcripción inversa.
Punto clave: la transcripción inversa es más importante para lograr la consistencia de la eficiencia de la transcripción inversa (las enzimas no solo deben ser eficientes sino también estables), en lugar de la cantidad de muestra cargada, si no es una PCR cuantitativa fluorescente a gran escala, no será posible en absoluto.Múltiples ADNc.
Varios fabricantes también han hecho algunos esfuerzos en la búsqueda de la consistencia.Por ejemplo, la mayoría de las empresas ahora han empaquetado la transcripción inversa como un kit estándar para la venta, lo cual es una buena opción.
Por ejemplo, los kits de la serie RT Easy de Foregene:
RT Easy I (premezcla maestra para el kit de síntesis de ADNc de la primera hebra)
MIQE (5) – información del gen diana

La figura anterior explica
1. Si este gen es efectivo para experimentos repetidos, generalmente se puede verificar mediante experimentos repetidos.
2. ID de gen, ya sabes.
3. Longitud del gen, la longitud total del gen objetivo definitivamente no es un problema.Al diseñar cebadores, asegúrese de que la longitud del amplicón esté entre 80 y 200 pb para garantizar una mejor eficiencia de amplificación.
4. Información de comparación de Sequence Blast, el gen objetivo debe compararse en el banco de genes para evitar la amplificación no específica.
5. Presencia de pseudogenes.Un pseudogen es una secuencia de ADN similar a un gen normal pero pierde su función normal.A menudo existe en la familia multigénica de eucariotas.Suele representarse por ψ.Es una copia de ADN genómico no funcional en el genoma que es muy similar a la secuencia del gen codificante., generalmente no se transcriben y no tienen un significado fisiológico claro.
6. Posición de los cebadores en relación con los exones e intrones.En los primeros años, cuando solucionamos el problema de la contaminación del ADN, a menudo prestamos atención a las posiciones de los cebadores, los exones y los intrones y, en general, consideramos diseñar cebadores a través de los intrones para evitar la amplificación del ADN.Consulte la siguiente figura: el negro representa los intrones, varios azules representan los exones, el rosa representa los cebadores comunes y el rojo brillante representa los cebadores que abarcan intrones.
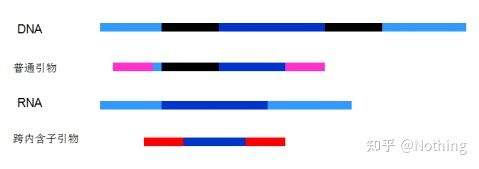
Esquemático, nunca cierto
Qué plan tan perfecto parece, pero de hecho, en la mayoría de los casos, los cebadores de trans-intrones no son tan mágicos como se imagina, y también causarán una amplificación no específica.Entonces, la mejor manera de prevenir la contaminación del ADN es eliminar el ADN por completo.
7. Predicción de la conformación.Usando este ejemplo nuevamente, use la herramienta en línea mFold para determinar la estructura secundaria del fragmento objetivo a una temperatura y concentración de sal específicas.
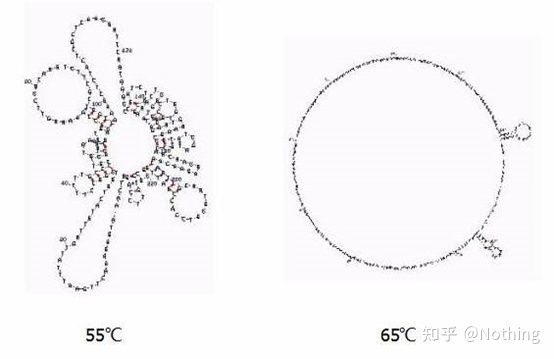
Estructura secundaria del ARN a diferentes temperaturas.
La estructura secundaria es el emparejamiento complementario de la propia plantilla, lo que conducirá a una fuerte competencia entre el cebador y el emparejamiento de plantilla, y las posibilidades de unión del cebador son menores, lo que da como resultado una serie de problemas como baja E y mala repetibilidad.A través de la predicción del software, si no hay un problema de estructura secundaria, sería genial.Si es así, nuestro artículo de seguimiento discutirá específicamente cómo resolver este problema.
MIQE (6): oligonucleótidos qPCR

Para la PCR cuantitativa fluorescente, lo primero con lo que lucha todos los días es la extracción de ARN, y lo segundo puede ser el diseño de cebadores.
En primer lugar, todavía verificamos las reglas sobre el diseño de imprimaciones de acuerdo con la lista de verificación MIQE.Es tan simple que los cabrones pueden reírse, y podemos terminarlo en una frase: averiguar la secuencia y la posición de la sonda cebadora y el método de modificación.Para el método de purificación de cebadores, la síntesis de cebadores es tan barata en la actualidad, qPCR es digno de PAGE y métodos de purificación superiores, y la información del instrumento de síntesis no es importante.Muchas personas llevan décadas haciendo cebadores y no saben que el sintetizador es ABI3900.
Con respecto a los principios del diseño de cartillas, no es necesario que los memorice de memoria, porque la mayoría de los programas de diseño de cartillas o herramientas en línea pueden resolver estos problemas (herramienta en línea recomendada primer3.ut.ee/), y el 99,999 % del diseño de cartillas no se hace manualmente. Mire, el autor a veces diseña cientos de cartillas al día, si lee uno por uno, se volverá bizco.
Simplemente verifique los siguientes puntos después de diseñar los cebadores:
1. Diseñe cebadores cerca del extremo 3': en el caso de usar cebadores oligo dT para la síntesis de la primera cadena de ADNc, teniendo en cuenta la eficiencia de la transcripción inversa y la integridad del ARN, los cebadores diseñados deben diseñarse cerca del extremo 3' para mejorar la eficiencia de amplificación.Use una imagen para explicar lo siguiente (no hay manera de entender esto):
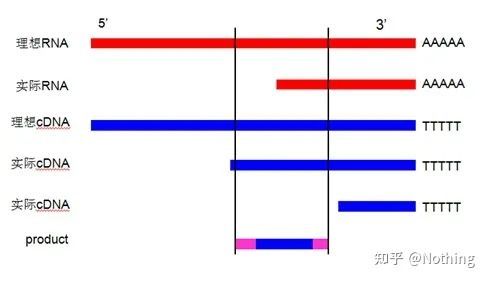
¿Por qué los cebadores deben diseñarse cerca del extremo 3 '? No debe ser cierto
2. Valor de TM: El valor de Tm está entre 55 y 65 °C (porque la actividad exonucleasa es máxima a 60 °C) y el contenido de GC está entre 40 % y 60 %.
3. BLAST: para evitar la amplificación no específica del genoma, se debe utilizar Blast para la verificación complementaria.
MIQE(7): proceso qPCR
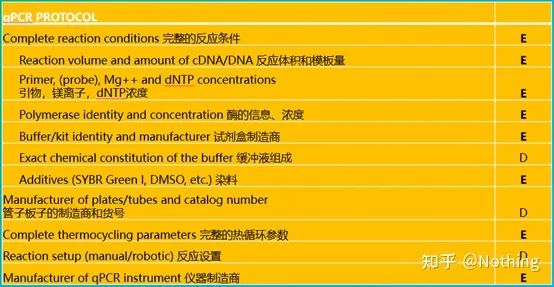
1. Kit qPCR
De acuerdo con los requisitos de MIQE, debemos describir claramente las condiciones de reacción completas en el artículo, incluida la configuración del sistema de reacción de PCR, qué kit se usa, quién es el fabricante, qué tan grande es el sistema de reacción, si se usa el método de colorante o el método de sonda, la configuración del programa de PCR.Los conductores veteranos definitivamente encontrarán que siempre que se seleccione el kit, la información anterior está básicamente determinada.
En la actualidad, la fabricación y producción de kits de PCR cuantitativa fluorescente es una tecnología muy madura.Mientras no elija fabricantes extremadamente malos, la probabilidad de problemas no es alta, pero aun así queremos compartir con usted algunos puntos:
Enzima Taq de arranque en caliente:La parte más importante de la PCR es la enzima Taq de arranque en caliente.Las enzimas de inicio en caliente en el mercado generalmente se dividen en dos tipos, una es una enzima de inicio en caliente modificada químicamente (puede imaginarla como inclusión en parafina) y la otra es una enzima de inicio en caliente para la modificación de anticuerpos (unión antígeno-anticuerpo).La modificación química es una forma temprana de enzimas de arranque en caliente.Cuando se alcanza cierta temperatura, la enzima liberará su actividad.La enzima de arranque en caliente modificada con anticuerpos utiliza métodos biológicos para bloquear la actividad de la enzima.Cuando se alcanza cierta temperatura, el anticuerpo se desnaturaliza y se inactiva como proteína, y la actividad enzimática entra en juego.
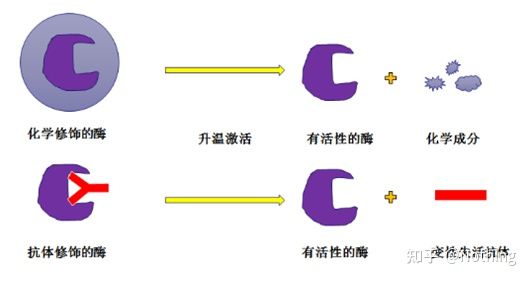
Sin embargo, ¿cuál es el uso de esto?Este es el caso, la actividad de liberación de las enzimas modificadas con anticuerpos es más rápida que la de las enzimas modificadas químicamente, por lo que en términos de sensibilidad, las enzimas modificadas con anticuerpos tienen una ligera ventaja, por lo que básicamente no hay enzimas modificadas químicamente en los kits del mercado.Si lo hay, entonces la tecnología de este fabricante todavía está atrapada en la era del milenio.
Concentración de iones de magnesio:La concentración de iones de magnesio es muy importante en la reacción de PCR.La concentración adecuada de iones de magnesio puede promover la liberación de la actividad de la enzima Taq.Si la concentración es demasiado baja, la actividad enzimática se reducirá significativamente;si la concentración es demasiado alta, se potenciará la amplificación no específica catalizada por enzimas.La concentración de iones de magnesio también afectará la hibridación de los cebadores, la temperatura de fusión de la plantilla y los productos de PCR, lo que afectará el rendimiento de los fragmentos amplificados.La concentración de iones de magnesio generalmente se controla a 25 mM.Por supuesto, para un buen kit, la concentración de iones de magnesio debe estar bien controlada.Algunos comerciantes agregan un agente quelante de iones de magnesio al reactivo, que puede lograr el efecto de ajuste automático de la concentración de iones de magnesio.
Concentración de colorante fluorescente:El colorante fluorescente, que es el SYBR Green que usamos habitualmente, genera principalmente fluorescencia al unirse al surco menor del ADN de doble cadena, porque la unión del colorante al ADN de doble cadena no es específica, es decir, siempre que se combine con él el ADN de doble cadena, puede producirse fluorescencia, por lo que los dímeros de cebador y las plantillas de ADN en el sistema se combinarán con él para formar una señal de fondo.
PD: Debido a sus propiedades sensibles a la luz, los productos en el mercado generalmente se envasan en tubos de centrífuga opacos de color marrón (como se muestra en la imagen a continuación).Sin embargo, esto encontrará un problema.Es difícil ver si el líquido es succionado cuando se toma una muestra.En este sentido, Qingke es de hecho el más fácil de usar (como se muestra en la imagen a continuación), y el tubo transparente está empacado en una bolsa de hojalata opaca.Luego póngalo en una bolsa de hojalata, teniendo en cuenta la conveniencia de evitar la luz y el muestreo.Debe elegir el número de producto correcto.TSE204 es una existencia súper rentable, lo que me da ganas de plantar césped.

La concentración del colorante fluorescente también es muy importante.Si la concentración es demasiado baja, la curva de amplificación no subirá en la etapa posterior y no será perfecta;si la concentración es demasiado alta, causará interferencia de ruido.Dado que la PCR cuantitativa fluorescente depende principalmente del valor de CT, si la concentración del colorante fluorescente no se ajusta correctamente, el punto bajo es mejor que el punto alto.Por supuesto, la concentración de tinte adecuada es la mejor.
ROX: Los colorantes ROX se utilizan para corregir los errores de señal de fluorescencia de pocillo a pocillo.Algunos fabricantes de instrumentos requieren calibración, mientras que otros no.Por ejemplo, el uso del instrumento de amplificación de PCR en tiempo real de Thermo Fisher Scientific generalmente requiere calibración, incluidos 7300, 7500, 7500Fast, StepOnePlus, etc. Las instrucciones generales del kit lo describirán.
La mezcla qPCR de Foregene también contiene tinte ROX, que es conveniente para usar en varios modelos.
Kit de PCR en tiempo real-Taqman
Tratamiento de enlace de hidrógeno débil: El tratamiento de los enlaces de hidrógeno débiles es un asunto relativamente técnico.Nada ha leído los manuales de muchos kits, pero ninguno menciona este tema.De hecho, es tan importante.La combinación de bases depende principalmente de la fuerza de los enlaces de hidrógeno.Los enlaces de hidrógeno fuertes son una amplificación normal y los enlaces de hidrógeno débiles conducen a una amplificación no específica.Si los enlaces de hidrógeno débiles no se pueden eliminar bien, no se puede evitar la amplificación no específica.Dentro del alcance del autor, solo unas pocas empresas han notado este problema.Cuando compre el kit, puede consultar si ha considerado una solución en este sentido para el kit que desea elegir.
Volumen de reacción: El sistema de 20-50 ul se usa con mayor frecuencia y es probable que los volúmenes más pequeños causen errores.En términos generales, las instrucciones del kit recomendarán el uso de volúmenes de reacción de PCR.No sea inteligente y use volúmenes más pequeños para ahorrar costos.el objetivo de.El volumen recomendado por los comerciantes en realidad ha sido probado, y es posible que no puedan resolver el problema de errores causados por pequeños volúmenes.
2. El fabricante y número de artículo de la placa de tubo
Todo el mundo conoce el principio de la PCR cuantitativa fluorescente.La recolección de fluorescencia se lleva a cabo principalmente a través de las tapas de los tubos de PCR.Al elegir consumibles para PCR, preste atención a dos puntos: buena transmisión de luz y adecuado para el instrumento.En términos generales, las placas y los tubos de las principales marcas están bien, pero debe elegir con cuidado en términos de adaptación, de lo contrario no podrá usar el instrumento.

4. Conocimiento de primer nivel
MIQE (8): validación qPCR
¡Esta es la máxima prioridad de qPCR!Tantos héroes han caído en la arena aquí.Por supuesto, también es posible que tengas suerte y los genes que estudiaste sean simples, por lo que flotaste a través de la cueva de hielo con el viento.La información de verificación de qPCR está destinada a probar la confiabilidad de los datos.Enumeramos la información de verificación necesaria de la siguiente manera:
1. Prueba de especificidad
La especificidad de la amplificación del gen diana se prueba comprobando si la imagen de la electroforesis es de una sola banda;verificación de secuenciación;curva de fusión para ver si el mapa de picos es único;verificación de la digestión enzimática y otros métodos.
Aquí, nos enfocamos en tEl análisis de la amplificación no específica por el método de las curvas de fusión.En términos generales, cuando diseñamos cebadores, se requiere que el tamaño del fragmento del producto esté en el rango de 80-200 pb, lo que hace que la temperatura de fusión del producto de PCR esté en el rango de 80-85 °C.Por lo tanto, si hay picos misceláneos, debe haber otros productos de amplificación no específicos;si el pico aparece por debajo de 80°C, generalmente se considera que es un dímero de cebador;si el pico aparece por encima de 85 °C, generalmente se considera que hay contaminación de ADN o más amplificación no específica de fragmentos grandes.
Nota: A veces solo hay un único pico a 80 °C.En este momento, este concepto debe ser respetado.Es probable que los resultados de la amplificación sean todos dímeros de cebadores.
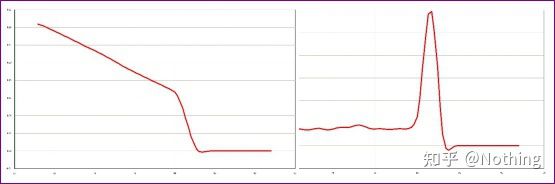
Curva de fusión normal (pico único sin amplificación no específica)
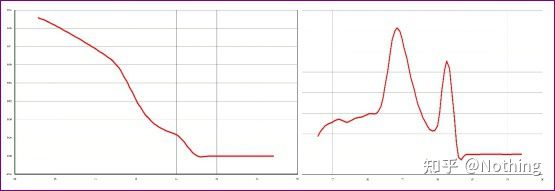
Curva de fusión problemática (amplificación no específica de picos espurios)
【Analisis de CASO】
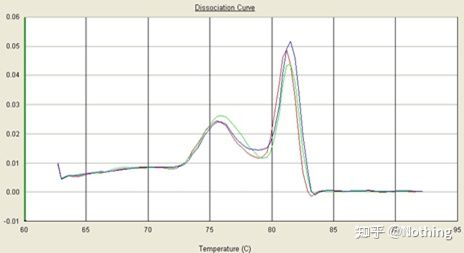
Hay un pico principal, pero el dímero del cebador es serio.
La curva de fusión de un solo pico en la figura a continuación puede engañar fácilmente a sus ojos, pensando que es un experimento perfecto, pero el resultado es completamente erróneo.En este momento, tenemos que fijarnos en la temperatura de fusión.La temperatura máxima está por debajo de 80°C, que es completamente dímero de cebador.
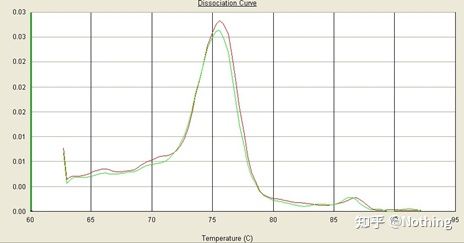
Sin fragmento objetivo, todos los dímeros de cebadores
Aquí, mi hermano no puede parar.La imagen de abajo es una foto tomada con un teléfono móvil que me envió un cabrón.Los reactivos que usó son todas marcas comúnmente usadas en la industria.Cambió de una marca de prefijo T a otra marca de prefijo T.Creo que ya lo has adivinado.El cabrón me gritó: “El reactivo utilizado en la primera imagen es demasiado bueno y el pico es único.Luego, después de usar el reactivo que recomendaste, se vuelve como la segunda imagen, con picos mixtos.Me has hecho miserable.“
Separa los dos gráficos.A primera vista, uno tiene un solo pico y el otro tiene un pico doble.Tonterías, un solo pico está bien, por supuesto.¿Es eso cierto?
Peor que Dou E, si pongo las dos imágenes en la imagen de abajo, lo entenderás de inmediato.De hecho, este tipo de imagen nos paraliza fácilmente.Después de un análisis cuidadoso, encontramos que: el pico de la primera figura está a 75°C, que es completamente dímero de cebador;el pico de la segunda cifra aparece a 75°C y 82°C, al menos hay El producto aparece.
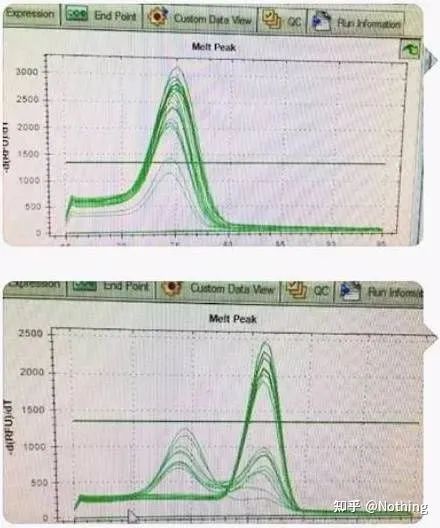
Imágenes de comentarios de los estudiantes.
Entonces, el problema fundamental no es el problema de los reactivos, sino el problema del diseño de cebadores.Al mismo tiempo, también prueba que algunas grandes marcas no son de calidad de hierro, y también prueba lo que dijo mi hermano antes: No es la marca de reactivo la que respalda su artículo.Es su artículo el que impulsó la marca de reactivos.Imagínese, si el cabrón no cambiara los reactivos, se enviarían datos incorrectos a la revista y lo que sucedería sería una tragedia.
2. Valor Ct del control en blanco
No explique, si el control en blanco tiene un valor Ct, ¿no es contaminación?Sin embargo, aún debe comprender qué control en blanco tiene un valor Ct.Si es NTC, significa que hay ADN extraño, como contaminación del reactivo.Si es NRT, significa que el ARN extraído tiene contaminación de ADN.
3. Curva estándar
Incluyendo la pendiente y la fórmula de cálculo, la eficiencia de PCR se puede calcular a través de la fórmula.Un experimento perfecto requiere que la pendiente de la curva estándar se acerque a 3,32 y que R² se acerque a 0,9999.
4. Rango dinámico lineal
El rango dinámico de la reacción es lineal.De acuerdo con la plantilla utilizada para generar la curva estándar, el rango dinámico debe incluir al menos 5 gradientes de concentración y prestar atención al cambio de los valores de Ct en gradientes de concentración altos y gradientes de concentración bajos.
5. Precisión de detección
Los cambios en los resultados de la qPCR, es decir, la mala repetibilidad, es decir, la mala precisión, se deben a muchos factores, como la temperatura, la concentración y el funcionamiento.La precisión de qPCR generalmente se vuelve menos controlable a medida que disminuye el número de copias.Idealmente, la variación dentro del experimento, esta variación técnica debe ser distinta de la variación biológica, y las réplicas biológicas pueden abordar directamente las diferencias estadísticas en los resultados de qPCR entre grupos o tratamientos.En particular, para los ensayos de diagnóstico, se debe informar la mejor precisión entre ensayos (repetibilidad) entre sitios y operadores.
6. Eficiencia de detección y LOD (en multiplex qPCR)
LOD es la concentración más baja del 95 % de las muestras positivas detectadas.En otras palabras, la concentración de LOD contenida dentro de un conjunto de réplicas de genes objetivo no debe exceder el 5 % de las reacciones fallidas.Al realizar un análisis de qPCR multiplex, especialmente para la detección simultánea de mutaciones puntuales o polimorfismos, la qPCR multiplex debe proporcionar evidencia de que la precisión de múltiples fragmentos de destino no se ve comprometida en el mismo tubo, detección múltiple y detección de tubo único La eficiencia y el LOD deben ser los mismos.Especialmente cuando se amplifican simultáneamente genes diana de alta concentración y genes diana de baja concentración, se debe prestar atención a este problema.
Problemas y solucionesEn términos generales, los problemas que se encuentran a menudo en la depuración de qPCR se centran en los siguientes aspectos:
·amplificación no específica
·Difícil elección de la concentración de imprimación y problemas con los dímeros de imprimación
·La temperatura de recocido es inexacta
·La estructura secundaria afecta la eficiencia de amplificación
amplificación no específica
amplificación no específicaocurre, generalmente se considera si el diseño del cebador no es adecuado, pero si no tiene prisa por cambiar los cebadores, primero puede probar los siguientes métodos (el principio también se adjunta):
·Aumente la temperatura de recocido: intente hacer que los enlaces de hidrógeno débiles no se puedan mantener;
·Acorte el tiempo de recocido y elongación: reduzca la posibilidad de enlaces de hidrógeno débiles;
·Reducir la concentración de cebadores: reduce la posibilidad de unión de cebadores redundantes y regiones no objetivo;
Baja eficiencia de amplificación
La situación opuesta a la amplificación no específica: baja eficiencia de amplificación, y las medidas para hacer frente a la baja eficiencia de amplificación son todo lo contrario:
·Prolongar el tiempo de recocido y elongación;
·Cambie a PCR de tres pasos y reduzca la temperatura de hibridación;
·Aumentar la concentración de primer;
Ps: Muchos estudiantes de posgrado nacidos en los años 90 no están dispuestos a estudiar cómo depurar experimentos y esperan que el kit pueda resolver el problema por completo (si desea ir a una empresa de reactivos para realizar investigación y desarrollo después de graduarse), de hecho, los fabricantes de reactivos también piensan de esta manera, espero que sea una tontería. Se puede usar cuando lo obtenga, por lo que los fabricantes de reactivos han dedicado un gran esfuerzo a resolver el problema de la amplificación no específica, incluida la introducción de factores de absorción de enlaces H débiles.Para resolver fácilmente el problema, los tontos todavía tienen que leer la introducción de la compañía de reactivos para ver si hay un factor que absorba los enlaces de hidrógeno débiles.
Difícil elección de la concentración del cebador y problemas con los dímeros de cebador
Método 1: En términos generales, las instrucciones del kit para qPCR tienen sistemas recomendados y concentraciones de cebador recomendadas.
Método 2: Depuración mediante el establecimiento del gradiente de concentración del cebador.La imagen de abajo es robada de una compañía para ilustrar.La siguiente figura muestra los resultados cuantitativos de fluorescencia realizados con tres gradientes de concentración de cebador (100 nM, 250 nM, 500 nM) y cuatro gradientes de concentración de plantilla (0,1 ng, 1 ng, 10 ng, 100 ng).El valor Ct de los resultados experimentales se representa de la siguiente manera:
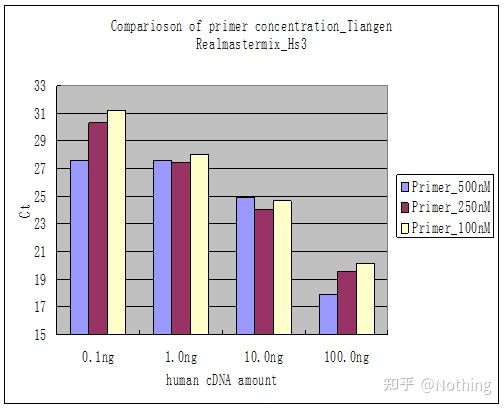
Selección de la concentración de cebador Concatenar cada concentración de cebador en una línea de la siguiente manera:
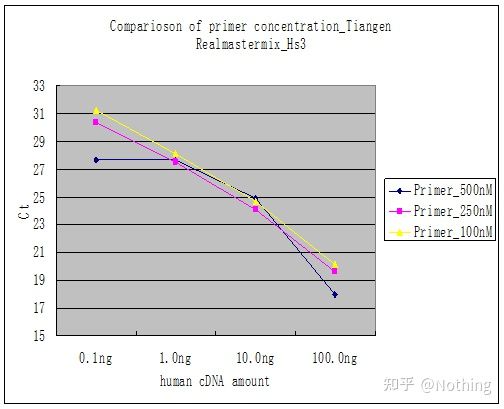
La elección de la concentración de cebador es obvia, la relación lineal de la concentración de cebador de 100 nM y 250 nM es mejor, y la relación lineal de la concentración de cebador de 500 nM es relativamente mala.En 100 nM y 250 nM, el valor de Ct de 250 nM es relativamente pequeño, por lo que la concentración de cebador óptima es 250 nM.En la curva de fusión pueden verse dímeros de imprimación generalmente severos.¿Qué pasa si los cebadores diseñados no pueden evitar los dímeros de cebadores?
Método 3: Reduzca la cantidad de cebadores y aumente la temperatura de recocido (no es necesario explicarlo).
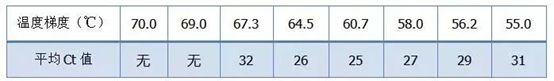
El valor empírico de la temperatura de recocido es de 60°C.Si no está seguro, ¿cómo seleccionar una temperatura de recocido más adecuada?La respuesta es la misma que la elección de la concentración de imprimación:prueba de gradiente.Tome una foto de la empresa Bio-rad para ilustrar el problema.Para la amplificación de un determinado fragmento diana, establezca ocho gradientes de temperatura, cada uno con tres repeticiones, y la curva de amplificación obtenida es la siguiente:
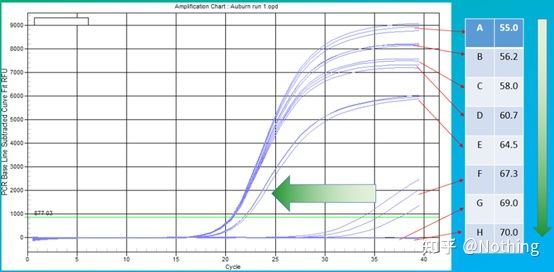
selección de la temperatura de recocido:
·70°C, 69°C: básicamente, los cebadores no se pueden combinar, por lo que no hay amplificación.
·67,3 °C: hay una pequeña cantidad de amplificación al principio y el valor de Ct es relativamente grande.
·64,5°C——El valor Ct disminuye.
·A 60,7 °C, 58,0 °C, 56,2 °C y 55,0 °C, los valores de Ct básicamente tendieron a ser estables, pero los valores finales de fluorescencia fueron diferentes.
¿Como escoger?Principio: El primer principio es el valor Ct más alto.Para el mismo valor de Ct, elija una temperatura de recocido más alta para evitar la dimerización y la amplificación no específica.Aunque hay un valor de fluorescencia más alto a 55 °C, puede haber dímeros o amplificación no específica en él.
Pero si eres tan inteligente como tú, definitivamente pensarás: Lógicamente hablando, si la reacción de PCR es muy específica, siempre que la concentración del cebador exceda el requisito mínimo, los puntos altos y bajos no deberían tener efecto, al igual que los colorantes fluorescentes y los dNTP.De hecho, siempre que la temperatura de recocido se optimice correctamente, el efecto de la concentración del cebador en el valor de Ct se minimizará naturalmente.
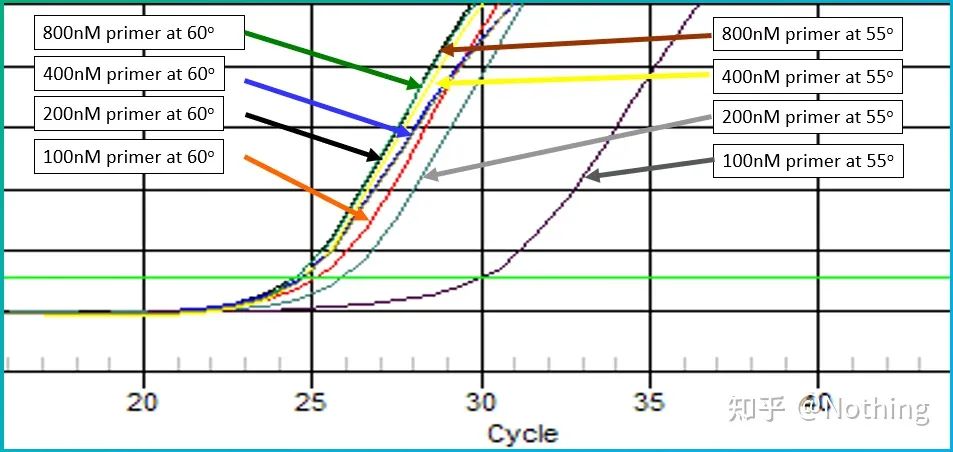
La temperatura de recocido se optimiza adecuadamente y el efecto de la concentración de imprimación en CT se minimizará
La estructura secundaria afecta la eficiencia de amplificación
Tomemos la foto de Bio-rad para ilustrar el problema.También diseña un gradiente de temperatura para amplificar un gen con una estructura secundaria.
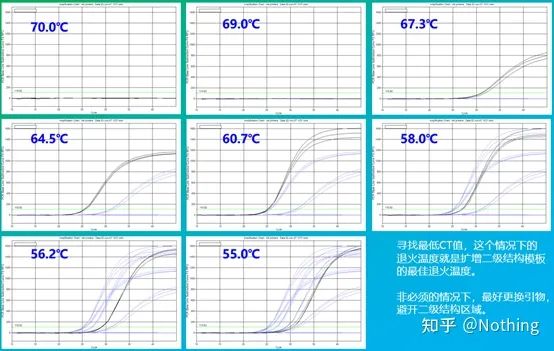
Emerge la estructura secundaria
Se puede observar que a medida que disminuye el gradiente de temperatura, comienzan a aparecer productos y el valor de Ct avanza, alcanzando el valor mínimo a 60,7°C, y luego a medida que disminuye el gradiente de temperatura, el valor de Ct aumenta.Por el contrario, a medida que aumenta la temperatura, la estructura secundaria se abre y aumenta la eficiencia de amplificación.Después de alcanzar cierta temperatura, aumentar la temperatura no puede mejorar la eficiencia de amplificación.Porque los cebadores no se pueden combinar de manera estable en este momento.Por lo tanto,busque la temperatura con el valor Ct más bajo, que es la mejor temperatura para amplificar la plantilla de la estructura secundaria.Por supuesto, los tontos inteligentes deben saber que si no es necesario, es mejor cambiar los cebadores y evitar la región de estructura secundaria.
5. Nivel de aplicación
MIQE—Análisis de datos

El análisis de datos se realiza principalmente mediante el instrumento PCR cuantitativo fluorescente.En el artículo anterior se ha hecho mucho trabajo de análisis de datos, como el control en blanco, que se ha explicado en el diseño del experimento.Se han aclarado los genes de referencia internos, el número de repeticiones, etc., aquí explicamos principalmente la aplicación de qPCR.
qPCR se usa ampliamente, y la verificación experimental y el diagnóstico de ácidos nucleicos son los escenarios más utilizados.
cuantificación absoluta
Log (concentración inicial) tiene una relación lineal con el número de ciclos.Se puede dibujar una curva estándar a partir de un estándar con un número de copias inicial conocido, es decir, se puede obtener la relación lineal de la reacción de amplificación.Según el valor Ct de la muestra, se puede calcular la concentración en la muestra.La cantidad de plantillas a incluir.
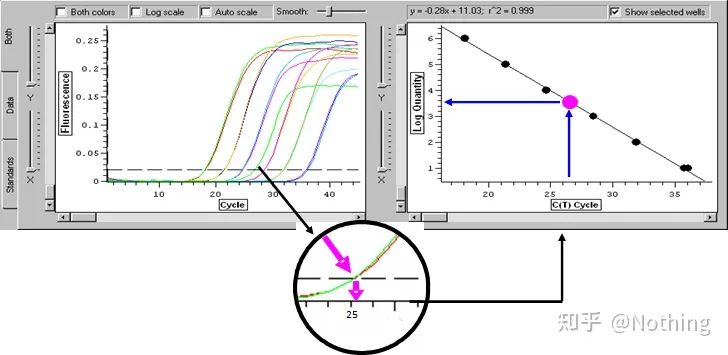
Método de cálculo cuantitativo absoluto
La cuantificación absoluta debe basarse en la curva estándar.Para hacer una curva estándar, se requiere un estándar.Por lo general, el estándar es un plásmido obtenido mediante la clonación del gen objetivo.¿Por qué es un plásmido?Porque el ADN plásmido circular es el más estable.Diluir el producto estándar en 5 a 6 gradientes según la proporción de duplicación (dilución de 10 veces) y prestar atención a la uniformidad al diluir.Deje que el valor de Ct caiga entre 15 y 30.
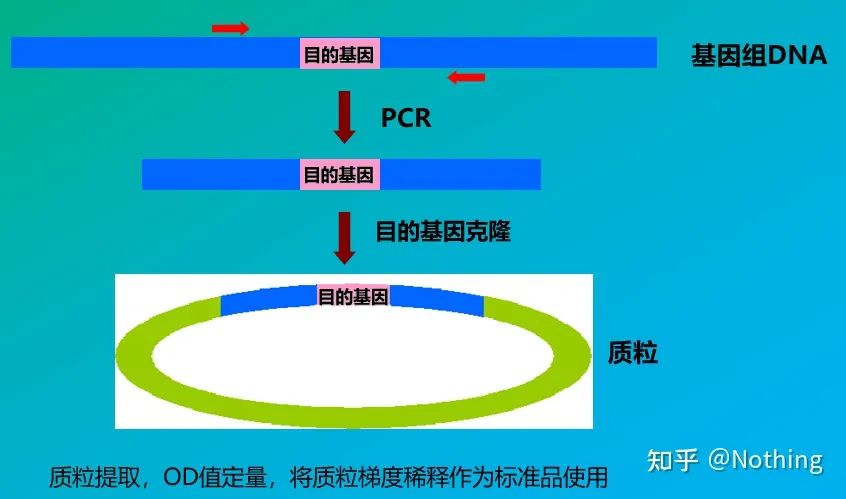
Preparación estándar
Al mismo tiempo, la muestra que se analizará también debe diluirse en consecuencia (recuerde el factor de dilución) y el valor Ct también debe estar entre 15 y 30.El producto estándar + la muestra a probar se colocan juntos en la máquina.Después de la corrida, se hizo una curva estándar con la sustancia estándar y las muestras a analizar se colocaron en la curva estándar para calcular la concentración.
La cuantificación del VHB del virus de la hepatitis B es una cuantificación absoluta típica, que puede calcular el número de copias del virus en 1 ml de sangre.
Cálculo del número de copias
Concentración de muestra a analizar (ng/ul) = OD260 × 50ug/ml × factor de dilución
Peso molecular de la muestra = número de bases × 324
El número de copias de la muestra a analizar (copias/ul) = la concentración de la muestra a analizar / el peso molecular de la muestra × 6 × 1014

Método de cálculo del número de copias
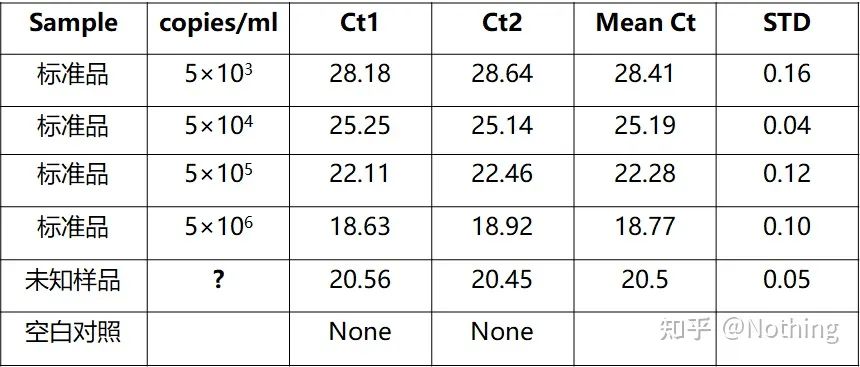

Lo anterior es el método de cálculo para determinar la cantidad.Este es un problema matemático que se puede resolver después de graduarse de la escuela secundaria y los problemas matemáticos generalmente se resuelven con computadoras.Si no entiendes, puedes venir a comunicarte.
cuantificación relativa
La cuantificación relativa se utiliza principalmente en la investigación científica.Cuántos virus hay en 1 ml de sangre, y es un virus de ADN, este es un evento relativamente determinista: se puede determinar la cantidad de sangre y el virus de ADN es relativamente estable.Sin embargo, es difícil para nosotros comparar el número de copias de transcripción de un determinado gen en una hoja, porque es difícil determinar el tamaño, el peso y la ternura de la hoja, la cantidad de ARN extraído es difícil de determinar y la eficiencia de la transcripción inversa también es difícil de determinar, es decir, cualquier paso puede hacer que los datos experimentales tengan errores y no se puedan usar.
Por lo tanto, la cuantificación relativa debe introducir un elemento:el gen de referencia interna.
En otras palabras, la cuantificación relativa es en realidad una comparación entre el gen objetivo y el gen de referencia interno.En comparación con el mismo tejido y la misma célula, la influencia del tamaño de la muestra, la cantidad de extracción de ARN, la eficiencia de la transcripción inversa y la eficiencia de la PCR es relativamente pequeña.Debido al pequeño tamaño de la muestra, tanto los genes de referencia internos como los genes diana se redujeron relativamente.Es por eso que hemos estado enfatizando la uniformidad y la estabilidad antes.
Los genes de referencia interna son generalmentegenes de limpieza(genes de mantenimiento), que se refieren a una clase de genes que se expresan de manera estable en todas las células, y sus productos son necesarios para mantener las actividades vitales básicas de las células.
No confundas este concepto.Los genes domésticos son términos de funciones biológicas, mientras que los genes de referencia internos son términos técnicos experimentales.Los genes domésticos deben pasar la validación antes de que puedan seleccionarse como genes de referencia internos.
Por ejemplo, seleccionamos varios genes domésticos en la figura a continuación para probar sus niveles de expresión en diferentes células de tejido y descubrimos que los niveles de expresión de β-2-microglobulina eran bastante diferentes de los de los otros tres genes, por lo que no podían usarse como genes de referencia internos.
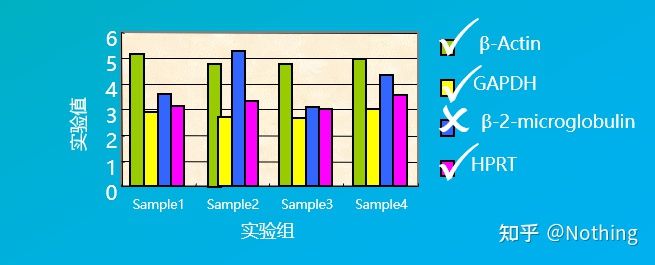
Después de comprender la función de corrección del gen de referencia interno, se derivan dos algoritmos debido a la introducción del gen de referencia interno.
·método de la doble curva estándar
·2 – Método △△Ct (método de comparación de valores CT)
Si está interesado en estudiar especies y funciones genéticas, abandone la investigación sobre algoritmos y use fórmulas directamente, o use máquinas directamente;Si eres un hombre heterosexual en matemáticas e ingeniería, siéntete libre.
método de doble curva estándar
Cuantifique el gen objetivo y el gen de limpieza de la muestra de control y la muestra que se analizará a través de la curva estándar, y luego calcule el valor relativo de acuerdo con la fórmula de cálculo, que es el nivel de expresión relativo.
Ventajas: análisis simple, optimización experimental relativamente simple
Desventaja: para cada gen, cada ronda de experimentos debe hacer una curva estándar
Aplicación: uno de los dos métodos cuantitativos relativos más utilizados y reconocidos en el estudio de la regulación de la expresión génica.
La fórmula es la siguiente:

Los ejemplos son los siguientes:
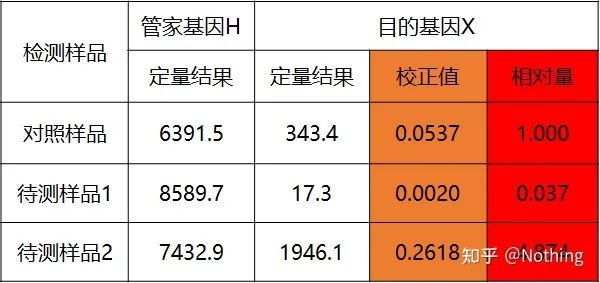
Calcular la cantidad relativa basada en el resultado cuantitativo
2 – Método △△Ct (método de comparación de valores CT)

Ventajas: No es necesario hacer una curva estándar
Desventajas: Se supone que la eficiencia de amplificación es cercana al 100%;la desviación estándar es < 5%, y se supone que la curva estándar y la eficiencia entre cada amplificación son consistentes;la optimización de las condiciones experimentales es más complicada.
Aplicación: uno de los dos métodos cuantitativos relativos más utilizados y reconocidos en el estudio de la regulación de la expresión génica.
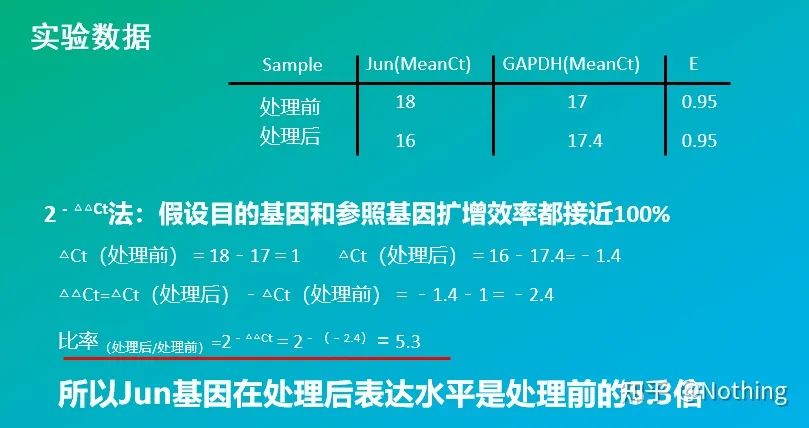
Por supuesto, la eficiencia de amplificación generalmente es imposible de ser perfectamente 1. Método de corrección: si sabemos que el gen objetivo y el gen de referencia tienen la misma eficiencia de amplificación, pero la eficiencia de amplificación no es igual a 1, entonces 2-△△Ct se puede corregir como: (1+E)-△△Ct, por ejemplo, si la eficiencia de amplificación es 0.95, entonces la fórmula de cálculo se puede corregir a 1.95- △△Ct
Hasta ahora, el contenido sobre la PCR cuantitativa fluorescente ha llegado a su fin.
Hora de publicación: 06-abr-2023



