



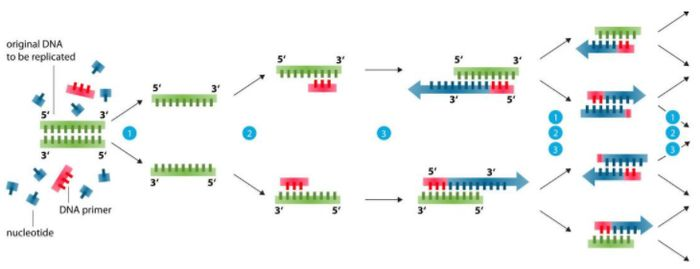
Base de diseño de imprimación (el 99% de los problemas se pueden resolver)
1. Longitud de la cartilla: el libro de texto requiere de 15 a 30 pb, generalmente alrededor de 20 pb.Es mejor que la condición real sea de 18 a 24 pb para garantizar la especificidad, pero cuanto más tiempo mejor, un cebador demasiado largo también reducirá la especificidad y reducirá el rendimiento.
2. Intervalo de amplificación del cebador: 200-500 pb es apropiado y el fragmento se puede expandir a 10 kb en condiciones específicas.
3. Base de imprimación: el contenido de G+C debe ser del 40-60%, muy poco efecto de amplificación de G+C no es bueno, demasiado G+C es fácil que aparezcan bandas no específicas.ATGC se distribuye mejor al azar, evitando grupos de más de 5 nucleótidos de purina o pirimidina.Multi-gc para el extremo 5′ y secuencias intermedias para aumentar la estabilidad, evitar GC rico en el extremo 3′, sin GC para las últimas 3 bases o sin GC para 3 de las últimas 5 bases.
4. Evite la estructura secundaria en los cebadores, y evite la complementación entre dos cebadores, especialmente la complementación en el extremo 3', de lo contrario se formará un dímero de cebador y se generarán bandas amplificadas no específicas.
5. Las bases en el extremo 3' de los cebadores, especialmente la última y la penúltima base, deben aparearse estrictamente para evitar fallas en la PCR debido a bases terminales no apareadas.
6. Los cebadores tienen o pueden agregarse con sitios de corte apropiados, y la secuencia diana amplificada debe tener preferiblemente sitios de corte apropiados, lo cual es muy beneficioso para el análisis de corte o la clonación molecular.
7. Especificidad de los cebadores: los cebadores no deben tener una homología obvia con otras secuencias en la base de datos de secuencias de ácidos nucleicos.
8. Aprenda a usar el software: PP5, Oligo6, DNAstar, Vector NTI, primer3 (este diseño en línea funciona mejor).
El contenido anterior puede resolver al menos el 99% de los problemas de diseño de cebadores.
Controle los detalles del diseño de imprimación
1. Longitud del cebador
La longitud general del cebador es de 18 a 30 bases.En general, el factor más importante que determina la temperatura de recocido de la imprimación es la longitud de la imprimación.La temperatura de recocido de la imprimación generalmente se selecciona (valor Tm -5 ℃), y algunos usan directamente el valor Tm.Las siguientes fórmulas se pueden utilizar para calcular aproximadamente la temperatura de recocido de las imprimaciones.
Cuando la longitud del cebador es inferior a 20 pb: [4(G+C)+2(A+T)]-5℃
Cuando la longitud del cebador es superior a 20 pb: 62,3 ℃+0,41 ℃ (%GC)-500/longitud-5 ℃
Además, también se pueden usar muchos programas para calcular la temperatura de recocido, el principio de cálculo será diferente, por lo que a veces el valor calculado puede tener una pequeña brecha.Para optimizar las reacciones de PCR, se utilizan los cebadores más cortos que aseguran temperaturas de hibridación de no menos de 54 ℃ para lograr la mejor eficiencia y especificidad.
En general, la especificidad del cebador aumenta en un factor de cuatro por cada nucleótido adicional, de modo que la longitud mínima del cebador para la mayoría de las aplicaciones es de 18 nucleótidos.El límite superior de la longitud del cebador no es muy importante, principalmente relacionado con la eficiencia de la reacción.Debido a la entropía, cuanto más largo sea el cebador, menor será la velocidad a la que se hibrida para unirse al ADN objetivo para formar una plantilla de doble cadena estable para que se una la ADN polimerasa.
Cuando se utiliza software para diseñar cebadores, la longitud de los cebadores se puede determinar a su vez por el valor de TM, especialmente para cebadores de PCR cuantitativa de fluorescencia, se debe controlar TM = 60 ℃ más o menos.
2.Contenido GC
En general, el contenido de G+C en las secuencias de cebadores es del 40 % al 60 %, y el contenido de GC y el valor de Tm de un par de cebadores deben coordinarse.Si el cebador tiene una tendencia A grave a GC o AT, se puede agregar la cantidad adecuada de cola A, T o G y C al extremo 5' del cebador.
3. Temperatura de recocido
La temperatura de recocido debe ser 5 ℃ más baja que la temperatura de desencadenamiento.Si el número de bases de cebador es pequeño, la temperatura de hibridación se puede aumentar adecuadamente, lo que puede aumentar la especificidad de la PCR.Si el número de bases es grande, la temperatura de recocido se puede reducir adecuadamente.La diferencia de temperatura de recocido entre un par de cebadores de 4 ℃ ~ 6 ℃ no afectará el rendimiento de la PCR, pero idealmente la temperatura de recocido de un par de cebadores es la misma, que puede variar entre 55 ℃ ~ 75 ℃.
4. Evite el área de estructura secundaria de la plantilla de amplificación
Es mejor evitar la región de estructura secundaria de la plantilla al seleccionar el fragmento amplificado.La estructura secundaria estable del fragmento de destino se puede predecir y estimar mediante un software informático relevante, que es útil para la selección de plantillas.Los resultados experimentales muestran que la expansión a menudo no tiene éxito cuando la energía libre (△G) de la región que se va a expandir es inferior a 58,6 lkJ/mol.
5. Falta de coincidencia con el ADN objetivo
Cuando la secuencia de ADN diana amplificada es grande, un cebador puede unirse a varias partes del ADN diana, lo que da como resultado la aparición de múltiples bandas en el resultado.Esta vez es necesario utilizar el sitio web de pruebas de software BLAST:http://www.ncbi.nlm.nih.gov/BLAST/.Seleccione Alinear dos secuencias (bl2seq).
Pegar secuencias de cebadores en la zona 1 y secuencias de ADN objetivo en la zona 2 es intercambiable, y BLAST calcula posibilidades complementarias, antisentido y otras, por lo que los usuarios no necesitan notar si ambas cadenas son cadenas con sentido.También puede ingresar el número de GI si conoce el número de GI de la secuencia en la base de datos, para que no tenga que pegar una gran parte de la secuencia.Finalmente, haga clic en Alinear en 3 para ver si el cebador tiene múltiples sitios homólogos en el ADN objetivo.
6. Terminal de imprimación
El extremo 3' del cebador es donde comienza la extensión, por lo que es importante evitar que los desajustes comiencen allí.El extremo 3' no debe tener más de 3 G o C consecutivos, porque esto hará que el cebador se active por error en la región de la secuencia de enriquecimiento G+C.El extremo 3' no puede formar ninguna estructura secundaria, excepto en reacciones especiales de PCR (AS-PCR), el extremo 3' del cebador no puede estar desapareado.Por ejemplo, si se amplifica la región codificante, el extremo 3' del cebador no debe terminar en la tercera posición del codón, porque la tercera posición del codón es propensa a degenerar, lo que afectará la especificidad y la eficiencia de la amplificación.Cuando use cebadores de anexión, consulte la tabla de uso de codones, preste atención a la preferencia biológica, no use cebadores de anexión en el extremo 3 'y use una concentración más alta de cebadores (1uM-3uM).
7. Estructura secundaria de los cebadores
Los cebadores en sí mismos no deben tener secuencias complementarias, de lo contrario, los cebadores en sí mismos se doblarán en estructuras de horquilla, y esta estructura secundaria afectará la unión de los cebadores y las plantillas debido al impedimento estérico.Si se utiliza el juicio artificial, las bases complementarias continuas de los propios cebadores no deben ser mayores de 3 pb.No debe haber complementariedad entre los dos cebadores, especialmente se debe evitar la superposición complementaria del extremo 3' para evitar la formación de dímeros de cebadores.En general, no debe haber más de 4 bases consecutivas de homología o complementariedad entre un par de cebadores.
8. Agregue marcadores o loci
El extremo 5' tiene poco efecto sobre la especificidad de amplificación y, por lo tanto, puede modificarse sin afectar la especificidad de amplificación.La modificación del extremo 5' del cebador incluyó: agregar el sitio de restricción de la enzima;Biotina marcada, fluorescencia, digoxina, Eu3+, etc. Introducir secuencias de ADN de unión a proteínas;Introducir sitios de mutación, insertar y perder secuencias de mutación e introducir secuencias promotoras, etc. Las bases adicionales afectarán más o menos la eficiencia de la amplificación y aumentarán la posibilidad de formación de dímeros de cebadores, pero se deben hacer algunas concesiones para el siguiente paso.Se pueden agregar secuencias adicionales que no existen en la secuencia objetivo, como sitios de restricción y secuencias promotoras, al extremo 5' del cebador sin afectar la especificidad.Estas secuencias no se incluyen en el cálculo de los valores de Tm de los cebadores, pero deben probarse en cuanto a complementariedad y estructura secundaria interna.
9. Subclones
La mayoría de las veces, la PCR es solo una clonación preliminar, y luego necesitamos subclonar el fragmento objetivo en varios vectores, por lo que necesitamos diseñar bases adicionales para la siguiente operación en el paso de la PCR.
A continuación se resumen algunas secuencias diseñadas para la subclonación.
Se agregó el sitio de restricción de la endonucleasa de restricción
La adición de sitios de restricción de enzimas es el método más utilizado para subclonar productos de PCR.En general, el sitio de escisión es de seis bases, además del extremo 5 'del sitio de escisión es necesario agregar 2 ~ 3 bases protectoras.Sin embargo, el número de bases protectoras requeridas por diferentes enzimas es diferente.Por ejemplo, SalⅠ no requiere base protectora, EcoRⅤ requiere 1 base protectora, NotⅠ requiere 2 bases protectoras y Hind Ⅲ requiere 3 bases protectoras.
LIC agrega la cola
El nombre completo de LIC es Clonación independiente de la ligadura, un método de clonación inventado por Navogen específicamente para su parte del vector pET.El portador de pET preparado por el método LIC tiene extremos cohesivos de una sola hebra de base 12-15 no complementarios, que complementan los extremos cohesivos correspondientes en el fragmento del inserto objetivo.Para fines de amplificación, la secuencia 5' del cebador del fragmento insertado debe complementar el vector LIC.La actividad extraída 3′→5′ de la ADN polimerasa T4 puede formar un extremo cohesivo monocatenario en el fragmento insertado después de un breve período de tiempo.Debido a que el producto solo puede formarse a partir de la hibridación mutua del fragmento de inserción preparado y el vector, este método es muy rápido y eficiente, y es una clonación dirigida.
Clon de TA dirigido agregar cola
La clonación de TA no pudo dirigir el fragmento a un vector, por lo que más tarde Invitrogen introdujo un vector que podía dirigirse a la clonación, que contenía cuatro GTGGS de base prominentes en un extremo.Por lo tanto, en el diseño de cebadores de PCR, las secuencias complementarias deben agregarse en consecuencia, para que los fragmentos puedan "orientarse".
Si tiene poco tiempo, puede intentar la síntesis directa, combinando el gen con el vector, que es lo que llamamos síntesis del gen ET en los musecularistas.
D. Método de clonación In-Fusion
No se requiere ligasa, no se requiere una reacción prolongada.Siempre que se introduzca una secuencia en ambos extremos del vector linealizado en el diseño de los cebadores, luego se agreguen el producto de la PCR y el vector linealizado a la solución enzimática de infusión que contiene BSA y se coloque a temperatura ambiente durante media hora, se puede realizar la transformación.Este método es particularmente adecuado para la conversión de grandes volúmenes.
10. Combinación de cartilla
A veces, solo se conoce información de secuencia limitada sobre el diseño de cebadores.Por ejemplo, si solo se conoce la secuencia de aminoácidos, se puede diseñar el cebador de fusión.Un cebador de fusión es una mezcla de diferentes secuencias que representan todas las diferentes posibilidades de bases que codifican un solo aminoácido.Para aumentar la especificidad, puede consultar la tabla de uso de codones para reducir la anexión según las preferencias de uso de bases de diferentes organismos.La hipoxantina se puede combinar con todas las bases para reducir la temperatura de recocido de la imprimación.No utilice las bases adjuntas en el extremo 3' del cebador porque la hibridación de las últimas 3 bases en el extremo 3' es suficiente para iniciar la PCR en el sitio equivocado.Se utilizan concentraciones de cebador más altas (1 μM a 3 μM) porque los cebadores en muchas mezclas de anexión no son específicos de la plantilla de destino.
Materias primas de PCRcontrol
1. Cantidad de imprimación
La concentración de cada cebador es de 0,1 ~ 1 umol o 10 ~ 100 pmol.Es mejor producir el resultado requerido con la menor cantidad de imprimación.Una alta concentración de cebador provocará un desajuste y una amplificación no específica, y aumentará la posibilidad de formar dímeros entre los cebadores.
2. Concentración de imprimación
La concentración de cebadores afecta la especificidad.La concentración de cebador óptima es generalmente entre 0,1 y 0,5 μM.Las concentraciones de cebador más altas conducen a la amplificación de productos no específicos.
3. Temperatura de recocido de la imprimación
Otro parámetro importante para las imprimaciones es la temperatura de fusión (Tm).Esta es la temperatura cuando el 50% de los cebadores y secuencias complementarias se representan como moléculas de ADN de doble cadena.Se requiere Tm para establecer la temperatura de hibridación de PCR.Idealmente, la temperatura de hibridación es lo suficientemente baja para asegurar la hibridación efectiva de los cebadores con la secuencia objetivo, pero lo suficientemente alta para reducir la unión no específica.Temperatura de recocido razonable de 55 ℃ a 70 ℃.La temperatura de recocido generalmente se establece 5 ℃ por debajo de la Tm de la imprimación.
Existen varias fórmulas para establecer la Tm, que varían mucho según la fórmula utilizada y la secuencia de los cebadores.Debido a que la mayoría de las fórmulas proporcionan un valor de Tm estimado, todas las temperaturas de recocido son solo un punto de partida.La especificidad se puede mejorar analizando varias reacciones que aumentan progresivamente la temperatura de recocido.Comience por debajo de la Tm-5 ℃ estimada y aumente gradualmente la temperatura de recocido en un incremento de 2 ℃.Una temperatura de recocido más alta reducirá la formación de dímeros de imprimación y productos no específicos.Para obtener los mejores resultados, los dos cebadores deben tener valores de Tm aproximados.Si la diferencia de Tm de los pares de cebadores es superior a 5 ℃, los cebadores mostrarán un inicio en falso significativo al usar una temperatura de recocido más baja en el ciclo.Si las Tm de los dos cebadores son diferentes, establezca la temperatura de recocido en 5 ℃ por debajo de la Tm más baja.Alternativamente, para aumentar la especificidad, se pueden realizar primero cinco ciclos a temperaturas de recocido diseñadas para Tm más altas, seguidos por los ciclos restantes a temperaturas de recocido diseñadas para Tm más bajas.Esto permite obtener una copia parcial de la plantilla de destino en condiciones estrictas.
4. Pureza y estabilidad de la imprimación
La pureza estándar de los cebadores personalizados es adecuada para la mayoría de las aplicaciones de PCR.La eliminación de los grupos benzoílo e isobutililo por desalinización es mínima y, por lo tanto, no interfiere con la PCR.Algunas aplicaciones requieren purificación para eliminar cualquier secuencia que no sea de longitud completa en el proceso de síntesis.Estas secuencias truncadas ocurren porque la eficiencia de la química de síntesis de ADN no es del 100%.Este es un proceso circular que utiliza reacciones químicas repetidas a medida que se agrega cada base para formar ADN de 3′ a 5′.Puedes fallar en cualquier ciclo.Los cebadores más largos, especialmente los que tienen más de 50 bases, tienen una gran proporción de secuencias truncadas y pueden requerir purificación.
El rendimiento de los cebadores se ve afectado por la eficiencia de la química sintética y el método de purificación.Las empresas biofarmacéuticas, como Cytology y Shengong, utilizan una unidad mínima de OD para garantizar la producción total de oligonucleósido.Los imprimadores personalizados se envían en forma de polvo seco.Lo mejor es volver a disolver los cebadores en TE para que la concentración final sea de 100 μM.TE es mejor que el agua desionizada porque el pH del agua suele ser ácido y provocará la hidrólisis de los oligonucleósidos.
La estabilidad de los primers depende de las condiciones de almacenamiento.El polvo seco y las imprimaciones disueltas deben almacenarse a -20 ℃.Los cebadores disueltos en TE en concentraciones superiores a 10 μM se pueden almacenar de forma estable a -20 ℃ durante 6 meses, pero solo se pueden almacenar a temperatura ambiente (15 ℃ a 30 ℃) durante menos de 1 semana.Las imprimaciones en polvo seco se pueden almacenar a -20 C durante al menos 1 año ya temperatura ambiente (15 C a 30 C) hasta 2 meses.
5. Enzimas y sus concentraciones
En la actualidad, la ADN polimerasa Taq utilizada es básicamente la enzima de ingeniería genética sintetizada por bacterias coliformes.La cantidad de enzima necesaria para catalizar una reacción de PCR típica es de aproximadamente 2,5 U (se refiere al volumen total de reacción de 100 ul).Si la concentración es demasiado alta, puede provocar una amplificación no específica;si la concentración es demasiado baja, se reducirá la cantidad de producto sintético.
6. Calidad y concentración de dNTP
La calidad de dNTP está estrechamente relacionada con la concentración y la eficiencia de la amplificación por PCR.El polvo de dNTP es granular y su variabilidad pierde su actividad biológica si no se almacena correctamente.La solución de dNTP es ácida y debe usarse en alta concentración, con solución tampón 1M NaOH o 1M Tris.HCL para ajustar su PH a 7.0 ~ 7.5, pequeña cantidad de subempaque, almacenamiento congelado a -20 ℃.La congelación-descongelación múltiple degradará el dNTP.En la reacción de PCR, el dNTP debe ser de 50 ~ 200 umol/L.Especialmente, se debe prestar atención a que la concentración de los cuatro DNTPS sea igual (preparación de igual mol).Si la concentración de alguno de ellos es diferente a la de los demás (mayor o menor), se producirá un desajuste.Una concentración demasiado baja reducirá el rendimiento de los productos de PCR.dNTP puede combinarse con Mg2+ y reducir la concentración de Mg2+ libre.
7. Ácido nucleico molde (gen diana)
La cantidad y el grado de purificación del ácido nucleico molde es uno de los vínculos clave para el éxito o el fracaso de la PCR.Los métodos tradicionales de purificación de ADN suelen utilizar SDS y proteasa K para digerir y desechar las muestras.Las funciones principales de SDS son: disolver lípidos y proteínas en la membrana celular, destruyendo así la membrana celular al disolver proteínas de membrana y disociar proteínas nucleares en la célula, SDS también puede combinarse con proteínas y precipitar;La proteasa K puede hidrolizar y digerir proteínas, especialmente histonas unidas con ADN, y luego usar fenol y cloroformo como solvente orgánico para extraer proteínas y otros componentes celulares, y usar etanol o alcohol isopropílico para precipitar el ácido nucleico.El ácido nucleico extraído se puede utilizar como molde para reacciones de PCR.Para muestras de detección clínica general, se puede usar un método rápido y simple para disolver células, lisar patógenos, digerir y eliminar proteínas de los cromosomas para liberar genes diana y usar directamente para la amplificación por PCR.La extracción de la plantilla de ARN generalmente utiliza el isotiocianato de guanidina o el método de la proteasa K para evitar que la RNasa degrade el ARN.
8. Concentración de Mg2+
Mg2+ tiene un efecto significativo sobre la especificidad y el rendimiento de la amplificación por PCR.En general, la reacción de PCR, cuando la concentración de varios dNTP es de 200 umol/L, la concentración adecuada de Mg2+ es de 1,5 ~ 2,0 mmol/L.La concentración de Mg2+ es demasiado alta, la especificidad de la reacción disminuye, se produce una amplificación no específica, una concentración demasiado baja reducirá la actividad de la ADN polimerasa Taq, lo que resultará en la reducción de los productos de reacción.
Los iones de magnesio afectan varios aspectos de la PCR, como la actividad de la ADN polimerasa, que afecta el rendimiento;Otro ejemplo es el recocido de cebadores, que afecta la especificidad.El dNTP y la plantilla se unen al ion de magnesio, lo que reduce la cantidad de ion de magnesio libre necesaria para la actividad enzimática.La concentración óptima de iones de magnesio varía para diferentes pares de cebadores y plantillas, pero una concentración inicial de PCR típica con dNTP 200 μM es de 1,5 mM (nota: para PCR cuantitativa en tiempo real, use una solución de iones de magnesio de 3 a 5 mM con una sonda fluorescente).Las concentraciones más altas de iones de magnesio libres aumentan el rendimiento, pero también aumentan la amplificación no específica y disminuyen la fidelidad.Para determinar la concentración óptima, se realizaron valoraciones de iones de magnesio en incrementos de 0,5 mM desde 1 mM a 3 mM.Para reducir la dependencia de la optimización de iones de magnesio, se puede utilizar la ADN polimerasa Platinum Taq.La ADN polimerasa Platinum Taq es capaz de mantener la función en un rango más amplio de concentraciones de iones de magnesio que la ADN polimerasa Taq y, por lo tanto, requiere menos optimización.
9. Aditivos promotores de pcr
La optimización de la temperatura de recocido, el diseño del cebador y la concentración de iones de magnesio es suficiente para la amplificación altamente específica de la mayoría de las plantillas;sin embargo, algunas plantillas, incluidas aquellas con alto contenido de GC, requieren medidas adicionales.Los aditivos que afectan la temperatura de fusión del ADN proporcionan otra forma de mejorar la especificidad y el rendimiento del producto.Se requiere la desnaturalización completa de la plantilla para obtener mejores resultados.
Además, la estructura secundaria evita la unión del cebador y la extensión de la enzima.
Los aditivos de PCR, incluidos formamida, DMSO, glicerina, betaína y solución potenciadora de PCRx, mejoran la amplificación.Su posible mecanismo es reducir la temperatura de fusión, lo que ayuda a la hibridación de los cebadores y ayuda a la extensión de la ADN polimerasa a través de la región de la estructura secundaria.PCRx Solution tiene otras ventajas.Se requiere una optimización mínima de iones de magnesio cuando se utiliza con ADN polimerasa Platinum Taq y ADN polimerasa Platinum Pfx.Por lo tanto, la técnica Platinum se combina con el aditivo para aumentar la especificidad y reducir la dependencia del tercer enfoque, la optimización de iones de magnesio.Para obtener mejores resultados, se debe optimizar la concentración de aditivos, especialmente DMSO, formamida y glicerol, que inhiben la ADN polimerasa Taq.

10. Arranque en caliente
La PCR de inicio en caliente es uno de los métodos más importantes para mejorar la especificidad de la PCR además de un buen diseño de cebadores.Aunque la temperatura óptima de elongación de la ADN polimerasa Taq es de 72 ℃, la polimerasa permanece activa a temperatura ambiente.Por tanto, se producen productos no específicos cuando la temperatura de mantenimiento es inferior a la temperatura de hibridación durante la preparación de la reacción de PCR y al comienzo del ciclo térmico.Una vez formados, estos productos no específicos se amplifican efectivamente.La PCR de inicio en caliente es particularmente eficaz cuando los sitios utilizados para el diseño de cebadores están limitados por la ubicación de los elementos genéticos, como mutaciones dirigidas al sitio, clonación de expresión o construcción y manipulación de elementos genéticos utilizados para la ingeniería de ADN.
Un método común para limitar la actividad de la ADN polimerasa Taq es preparar la solución de reacción de PCR en hielo y colocarla en un aparato de PCR precalentado.Este método es simple y económico, pero no completa la actividad de la enzima y, por lo tanto, no elimina por completo la amplificación de productos no específicos.
El cebado térmico retrasa la síntesis de ADN al inhibir un componente esencial hasta que el aparato de PCR alcanza la temperatura de desnaturalización.La mayoría de los métodos manuales de iniciación térmica, incluida la adición retardada de polimerasa de ADN Taq, son engorrosos, especialmente para aplicaciones de alto rendimiento.Otros métodos de cebado térmico utilizan un escudo de cera para encerrar un componente esencial, incluidos iones de magnesio o enzimas, o para aislar físicamente componentes reactivos, como plantillas y tampones.Durante el ciclo térmico, los diversos componentes se liberan y se mezclan a medida que la cera se derrite.Al igual que el método manual de arranque en caliente, el método de escudo de cera es engorroso y propenso a la contaminación y no es adecuado para aplicaciones de alto rendimiento.
La ADN polimerasa de platino es conveniente y eficiente para la PCR automática de inicio en caliente.La ADN polimerasa Platinum Taq consta de ADN polimerasa Taq recombinante combinada con un anticuerpo monoclonal contra la ADN polimerasa Taq.Los anticuerpos se formulan mediante PCR para inhibir la actividad enzimática durante el mantenimiento prolongado de la temperatura.La ADN polimerasa Taq se liberó en la reacción durante el aislamiento de 94 ℃ del paso de desnaturalización, restaurando la actividad polimerasa completa.A diferencia de la ADN polimerasa Taq modificada químicamente para la iniciación térmica, la enzima Platinum no requiere un aislamiento prolongado a 94 ℃ (10 a 15 minutos) para activar la polimerasa.Con la ADN polimerasa PlatinumTaq, el 90 % de la actividad de la ADN polimerasa Taq se restauró después de 2 minutos a 94 ℃.

11. Nido-PCR
Las rondas sucesivas de amplificación con cebadores anidados pueden mejorar la especificidad y la sensibilidad.La primera ronda es una amplificación estándar de 15 a 20 ciclos.Una pequeña fracción del producto de amplificación inicial se diluyó de 100 a 1000 veces y se agregó a la segunda ronda de amplificación durante 15 a 20 ciclos.Alternativamente, el producto amplificado inicial puede clasificarse por tamaño mediante purificación en gel.Se utiliza un cebador anidado en la segunda ronda de amplificación, que puede unirse a la secuencia objetivo dentro del primer cebador.El uso de PCR anidada reduce la posibilidad de amplificación de múltiples sitios objetivo porque hay pocas secuencias objetivo complementarias a ambos conjuntos de cebadores.El mismo número total de ciclos (30 a 40) con los mismos cebadores amplificaron los sitios no específicos.La PCR anidada aumenta la sensibilidad de las secuencias diana limitadas (p. ej., mrnas raros) y mejora la especificidad de las PCRS difíciles (p. ej., 5′ RACE).
12. PCR descendente
La PCR descendente mejora la especificidad mediante el uso de condiciones de hibridación estrictas para los primeros ciclos de PCR.El ciclo comienza a una temperatura de recocido aproximadamente 5 ℃ más alta que la Tm estimada, luego cada ciclo se reduce de 1 ℃ a 2 ℃ hasta que la temperatura de recocido está por debajo de Tm 5 ℃.Solo se ampliará la plantilla de destino con la mayor homología.Estos productos continúan expandiéndose en ciclos posteriores, desplazando a los productos no específicos amplificados.La PCR descendente es útil para los métodos en los que se desconoce el grado de homología entre el cebador y la plantilla de destino, como la toma de huellas dactilares de ADN AFLP.
Kits de PCR relacionados
El héroe 2× PCRTMEl sistema Mix tiene una mayor tolerancia a los inhibidores de PCR que el sistema PCR Mix normal y puede hacer frente fácilmente a la amplificación por PCR de varias plantillas complejas.El exclusivo sistema de reacción y Taq Hero de alta eficiencia hacen que la reacción de PCR tenga una mayor eficiencia de amplificación, especificidad y sensibilidad.
Mayor eficiencia de amplificación
Tiene actividad ADN polimerasa 5'→3' y actividad exonucleasa 5'→3', sin actividad exonucleasa 3'→5'.
PCR en tiempo real Easyᵀᴹ-SYBR Green I Kit
Específico: el tampón optimizado y la enzima Taq de arranque en caliente pueden prevenir la amplificación no específica y la formación de dímeros de cebadores
Alta sensibilidad: puede detectar copias bajas de la plantilla
El kit utiliza un reactivo de transcripción inversa único de Foregene y la polimerasa de ADN Foregene HotStar Taq combinadas con un sistema de reacción único para mejorar eficazmente la eficiencia de amplificación y la especificidad de la reacción.
Hora de publicación: 09-may-2023



