



RT-qPCR se desarrolla a partir de la tecnología PCR ordinaria.Añade productos químicos fluorescentes (tintes fluorescentes o sondas fluorescentes) al sistema de reacción de PCR tradicional y detecta el proceso de hibridación y extensión de PCR en tiempo real según sus diferentes mecanismos luminiscentes.Los cambios de señal fluorescente en el medio se utilizan para calcular la cantidad de cambio de producto en cada ciclo de PCR.Actualmente, los métodos más comunes son el método de colorante fluorescente y el método de sonda.
Método de tinte fluorescente:
Algunos tintes fluorescentes, como SYBR Green Ⅰ, PicoGreen, BEBO, etc., no emiten luz por sí mismos, sino que emiten fluorescencia después de unirse al surco menor de dsDNA.Por lo tanto, al comienzo de la reacción de PCR, la máquina no puede detectar la señal fluorescente.Cuando la reacción pasa a la etapa de hibridación-extensión (método de dos pasos) o etapa de extensión (método de tres pasos), las cadenas dobles se abren en este momento y la nueva ADN polimerasa Durante la síntesis de la cadena, las moléculas fluorescentes se combinan en el surco menor de dsDNA y emiten fluorescencia.A medida que aumenta el número de ciclos de PCR, más y más tintes se combinan con dsDNA, y la señal fluorescente también se mejora continuamente.Tome SYBR Green Ⅰ como ejemplo.
Método de sonda:
La sonda Taqman es la sonda de hidrólisis más utilizada.Hay un grupo fluorescente en el extremo 5 'de la sonda, generalmente FAM.La sonda en sí es una secuencia complementaria al gen diana.Hay un grupo de extinción fluorescente en el extremo 3 'del fluoróforo.De acuerdo con el principio de transferencia de energía de resonancia de fluorescencia (transferencia de energía de resonancia de Förster, FRET), cuando el grupo fluorescente reportero (molécula fluorescente donante) y el grupo fluorescente de extinción (molécula fluorescente aceptora) Cuando el espectro de excitación se superpone y la distancia es muy cercana (7-10nm), la excitación de la molécula donante puede inducir la fluorescencia de la molécula aceptora, mientras que la autofluorescencia se debilita.Por lo tanto, al comienzo de la reacción de PCR, cuando la sonda está libre e intacta en el sistema, el grupo fluorescente informador no emitirá fluorescencia.Al recocer, el cebador y la sonda se unen a la plantilla.Durante la etapa de extensión, la polimerasa sintetiza continuamente nuevas cadenas.La ADN polimerasa tiene actividad de exonucleasa 5′-3′.Al llegar a la sonda, la ADN polimerasa hidrolizará la sonda de la plantilla, separará el grupo fluorescente informador del grupo fluorescente extintor y liberará la señal fluorescente.Dado que existe una relación de uno a uno entre la sonda y la plantilla, el método de la sonda es superior al método de tinción en términos de precisión y sensibilidad de la prueba.
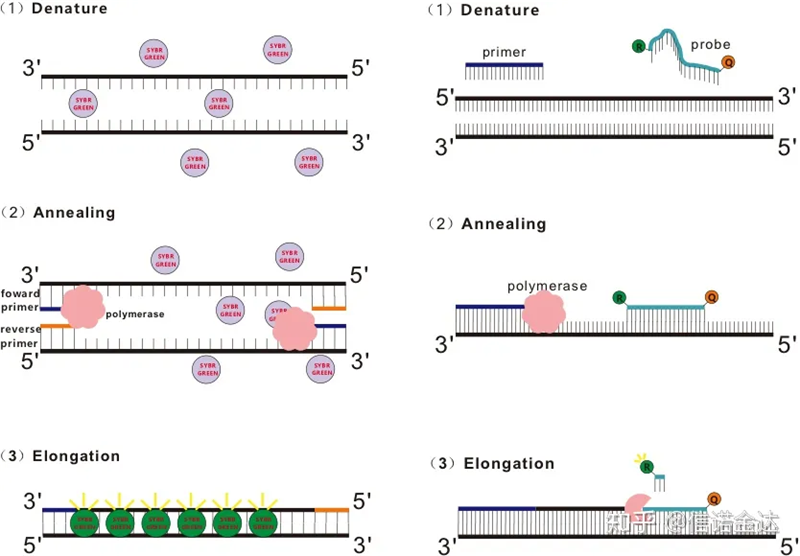
Fig. 1 Principio de qRT-PCR
Primer diseño
Principios:
Los cebadores deben diseñarse en la región conservada de la serie de ácidos nucleicos y tener especificidad.
Es mejor usar la secuencia de cDNA, y la secuencia de mRNA también es aceptable.Si no, averigüe el diseño de la región cds de la secuencia de ADN.
La longitud del producto cuantitativo fluorescente es de 80 a 150 pb, la más larga es de 300 pb, la longitud del cebador generalmente está entre 17 y 25 bases, y la diferencia entre los cebadores aguas arriba y aguas abajo no debe ser demasiado grande.
El contenido de G+C está entre 40% y 60%, y 45-55% es el mejor.
El valor de TM está entre 58-62 grados.
Trate de evitar dímeros y autodímeros de cebadores, (no aparezcan más de 4 pares de bases complementarias consecutivas) estructura de horquilla, si es inevitable, haga ΔG<4.5kJ/mol* Si no puede asegurar que el gDNA se haya eliminado durante la transcripción inversa Limpie, es mejor diseñar los cebadores del extremo del intrón * 3 'no se puede modificar, y para evitar las regiones ricas en AT, GC, evite los cebadores de estructura continua T/C, A/G (2-3) y no-
específica La homología de la secuencia heterogéneamente amplificada es preferentemente inferior al 70% o tiene una homología de 8 bases complementarias.
Base de datos:
CottonFGD búsqueda por palabras clave
Primer diseño:
Diseño de cebadores IDT-qPCR
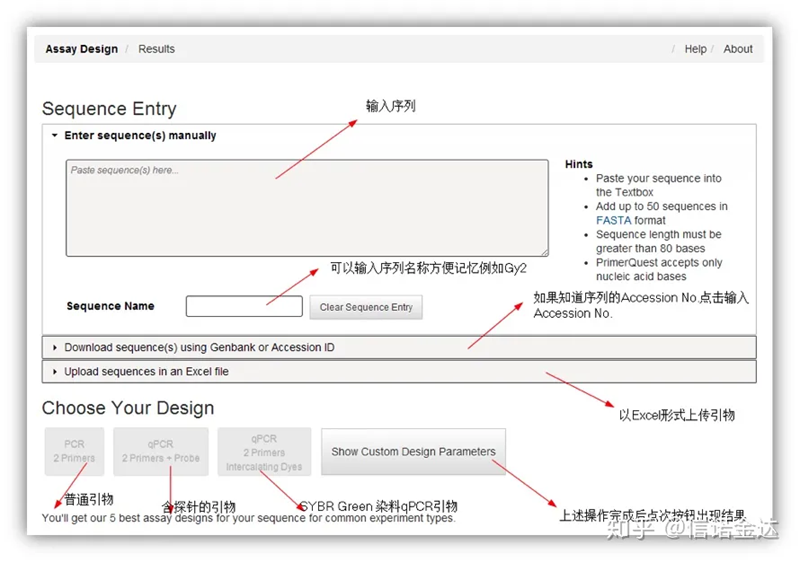
Página de la herramienta de diseño de cebadores en línea Fig2 IDT
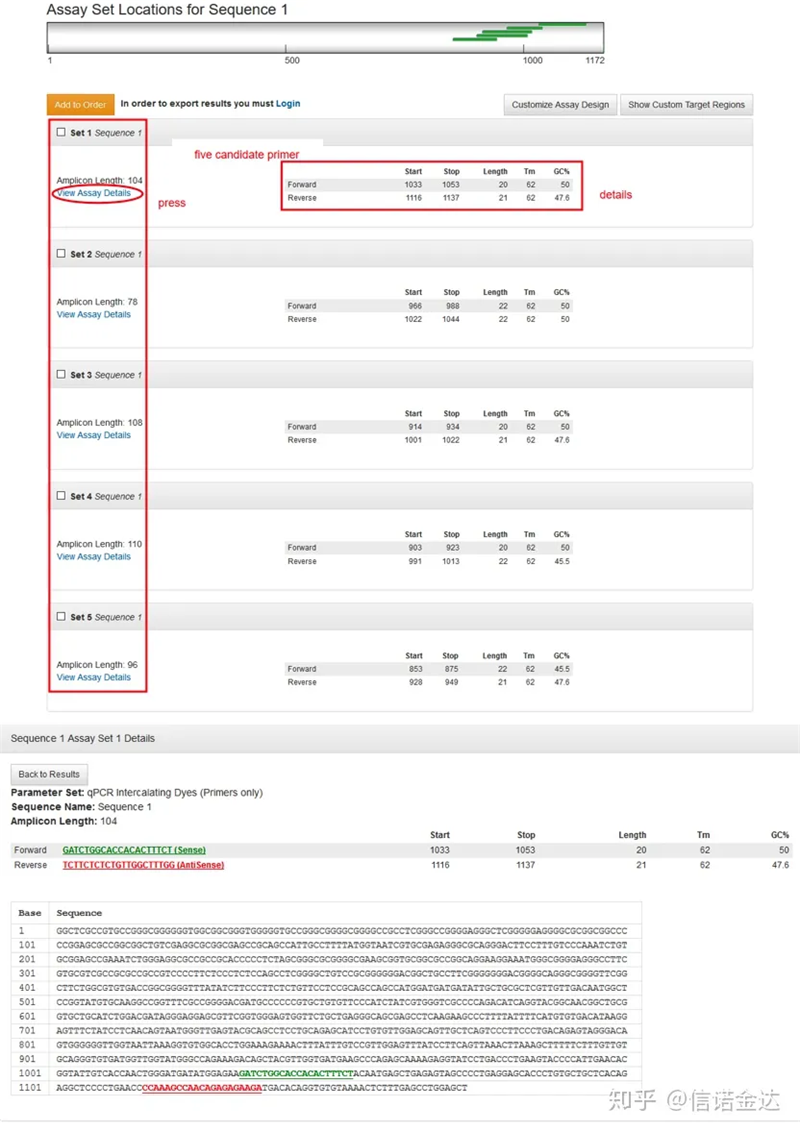
Visualización de la página de resultados Fig3
Diseño de cebadores lncRNA:
ARNlnc:los mismos pasos que el ARNm.
miARN:El principio del método stem-loop: dado que todos los miRNA son secuencias cortas de aproximadamente 23 nt, no se puede realizar la detección directa por PCR, por lo que se utiliza la herramienta de secuencia stem-loop.La secuencia de tallo-bucle es un ADN monocatenario de aproximadamente 50 nt, que puede formar una estructura de horquilla por sí mismo.3 'El final se puede diseñar como una secuencia complementaria al fragmento parcial de miARN, luego el miARN objetivo se puede conectar a la secuencia de bucle de tallo durante la transcripción inversa, y la longitud total puede alcanzar los 70 pb, que está en línea con la longitud del producto amplificado determinado por qPCR.Diseño de cebadores de miARN de cola.
Detección específica de amplificación:
Base de datos de explosión en línea: explosión de CottonFGD por similitud de secuencia
Explosión local: Consulte el uso de Blast+ para realizar una explosión local, linux y macos pueden establecer directamente una base de datos local, el sistema win10 también se puede realizar después de instalar ubuntu bash.Crear base de datos de voladuras locales y voladuras locales;abrir ubuntu bash en win10.
Aviso: el algodón Upland y el algodón Sea Island son cultivos tetraploides, por lo que el resultado de la explosión suele ser dos o más coincidencias.En el pasado, al usar cds de NAU como base de datos para realizar blast, es probable que se encontraran dos genes homólogos con solo unas pocas diferencias de SNP.Por lo general, los dos genes homólogos no se pueden separar mediante el diseño de cebadores, por lo que se tratan como iguales.Si hay un indel obvio, el cebador generalmente se diseña en el indel, pero esto puede conducir a la estructura secundaria del cebador. La energía libre aumenta, lo que lleva a una disminución en la eficiencia de amplificación, pero esto es inevitable.
Detección de la estructura secundaria del cebador:
Pasos:abra oligo 7 → ingrese la secuencia de la plantilla → cierre la subventana → guarde → ubique el cebador en la plantilla, presione ctrl+D para establecer la longitud del cebador → analice varias estructuras secundarias, como el cuerpo de autodimerización, el heterodímero, la horquilla, la falta de coincidencia, etc. Las dos últimas imágenes en la Figura 4 son los resultados de las pruebas de los cebadores.El resultado de la imprimación frontal es bueno, no hay una estructura obvia de dímero y horquilla, no hay bases complementarias continuas, y el valor absoluto de la energía libre es inferior a 4.5, mientras que la imprimación posterior muestra continuas Las 6 bases son complementarias y la energía libre es 8.8;además, aparece un dímero más serio en el extremo 3′ y aparece un dímero de 4 bases consecutivas.Aunque la energía libre no es alta, el dímero 3' Chl puede afectar seriamente la especificidad de amplificación y la eficiencia de amplificación.Además, es necesario verificar horquillas, heterodímeros y desajustes.
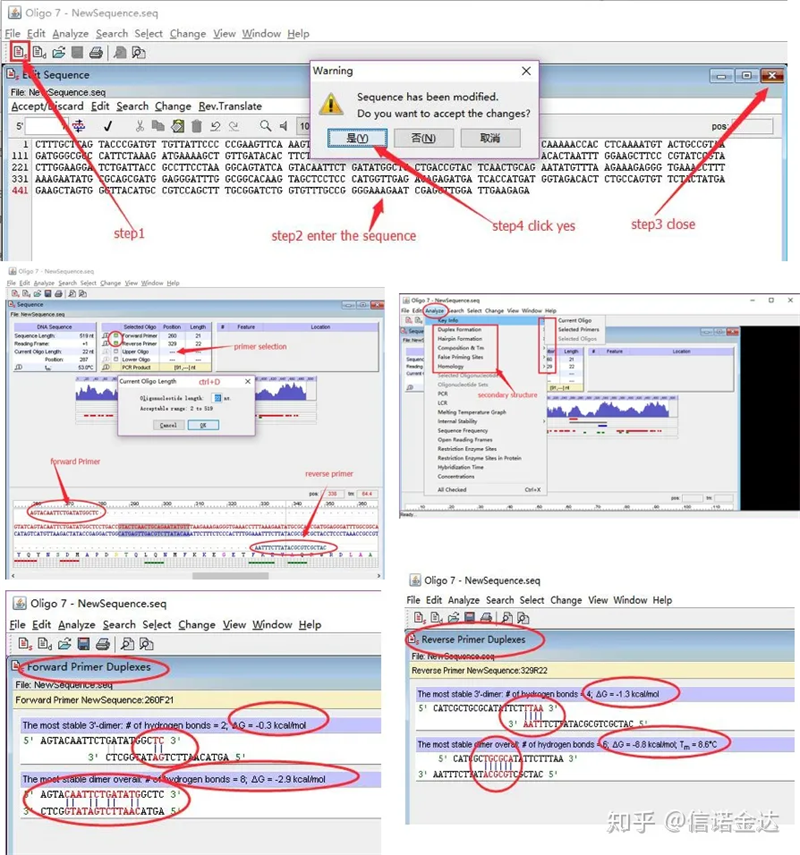
Resultados de detección de Fig3 oligo7
Detección de eficiencia de amplificación:
La eficiencia de amplificación de la reacción de PCR afecta seriamente los resultados de la PCR.También en qRT-PCR, la eficiencia de amplificación es particularmente importante para los resultados cuantitativos.Eliminar otras sustancias, máquinas y protocolos en el tampón de reacción.La calidad de los cebadores también tiene una gran influencia en la eficiencia de amplificación de qRT-PCR.Para garantizar la precisión de los resultados, tanto la cuantificación de fluorescencia relativa como la cuantificación de fluorescencia absoluta necesitan detectar la eficiencia de amplificación de los cebadores.Se reconoce que la eficiencia efectiva de amplificación de qRT-PCR está entre 85% y 115%.Hay dos métodos:
1. Método de la curva estándar:
a.Mezclar ADNc
b.Dilución de gradiente
c.qPCR
d.Ecuación de regresión lineal para calcular la eficiencia de amplificación
2. LinReg PCR
LinRegPCR es un programa para el análisis de datos de RT-PCR en tiempo real, también llamados datos de PCR cuantitativos (qPCR) basados en SYBR Green o química similar.El programa utiliza datos corregidos que no son de referencia, realiza una corrección de referencia en cada muestra por separado, determina una ventana de linealidad y luego utiliza un análisis de regresión lineal para ajustar una línea recta a través del conjunto de datos de PCR.A partir de la pendiente de esta línea se calcula la eficacia de la PCR de cada muestra individual.La eficacia media de la PCR por amplicón y el valor de Ct por muestra se utilizan para calcular una concentración inicial por muestra, expresada en unidades de fluorescencia arbitrarias.La entrada y salida de datos se realiza a través de una hoja de cálculo de Excel.solo muestra
se requiere mezclar, sin gradiente
se requieren pasos:(Tome Bole CFX96 como ejemplo, no exactamente Máquina con ABI claro)
experimento:es un experimento qPCR estándar.
Salida de datos qPCR:LinRegPCR puede reconocer dos formas de archivos de salida: RDML o resultado de amplificación de cuantificación.De hecho, es el valor de detección en tiempo real del número de ciclo y la señal de fluorescencia de la máquina, y la amplificación se obtiene analizando el valor de cambio de fluorescencia de la eficiencia del segmento lineal.
Selección de datos: En teoría, el valor RDML debería ser utilizable.Se estima que el problema de mi computadora es que el software no puede reconocer RDML, por lo que tengo el valor de salida de Excel como los datos originales.Se recomienda realizar primero una evaluación aproximada de los datos, como la falla al agregar muestras, etc. Los puntos se pueden eliminar en los datos de salida (por supuesto, no puede eliminarlos, LinRegPCR ignorará estos puntos en la etapa posterior)
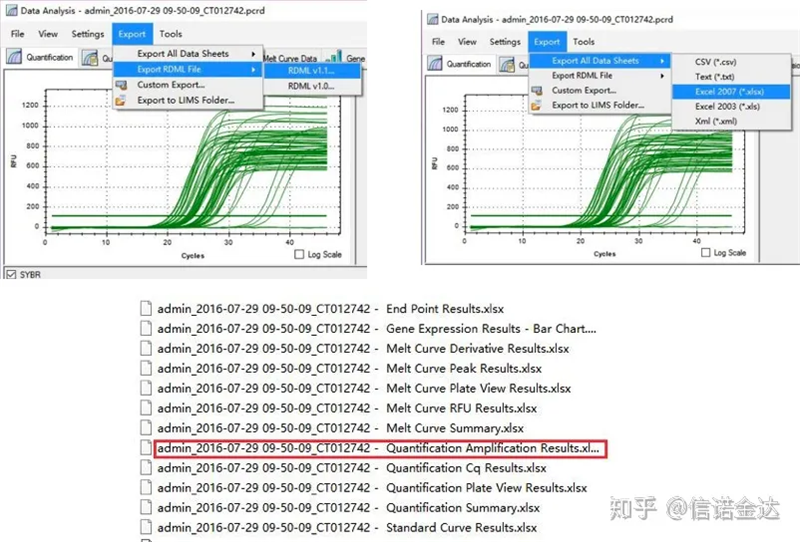
Exportación de datos de Fig5 qPCR
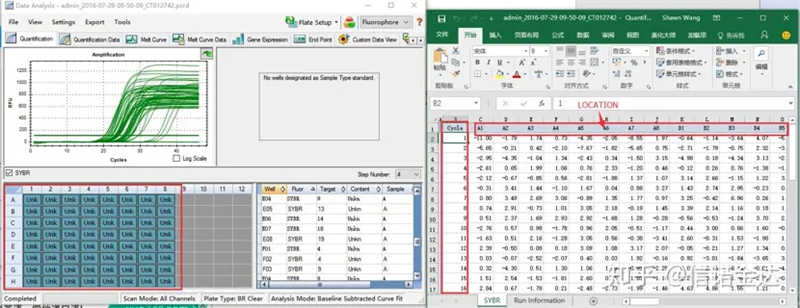
Fig6 selección de muestras candidatas
Entrada de datos:Abra los resultados de amplificación de calificación.xls, → abra LinRegPCR → archivo → lea desde Excel → seleccione los parámetros como se muestra en la Figura 7 → Aceptar → haga clic en determinar líneas de base
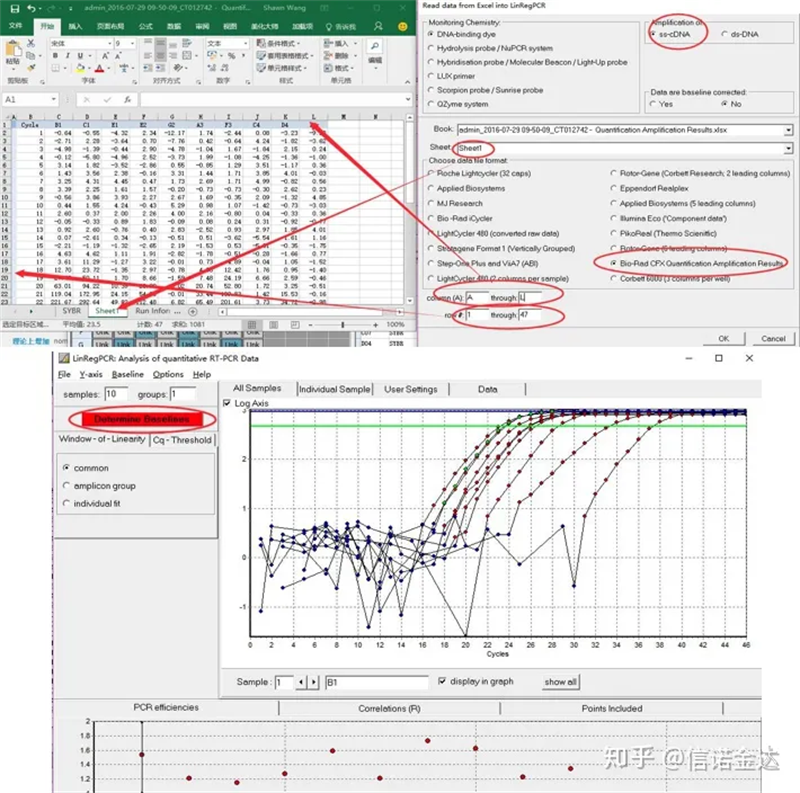
Fig. 7 pasos de entrada de datos linRegPCR
Resultado:Si no hay repetición, no se requiere agrupación.Si hay repetición, se puede editar la agrupación en la agrupación de muestra, y se ingresa el nombre del gen en el identificador, y luego se agrupará automáticamente el mismo gen.Finalmente, haga clic en el archivo, exporte Excel y vea los resultados.Se mostrarán la eficiencia de amplificación y los resultados de R2 de cada pocillo.En segundo lugar, si se divide en grupos, se mostrará la eficiencia de amplificación promedio corregida.Asegúrese de que la eficiencia de amplificación de cada cebador esté entre el 85 % y el 115 %.Si es demasiado grande o demasiado pequeño, significa que la eficiencia de amplificación del cebador es baja.
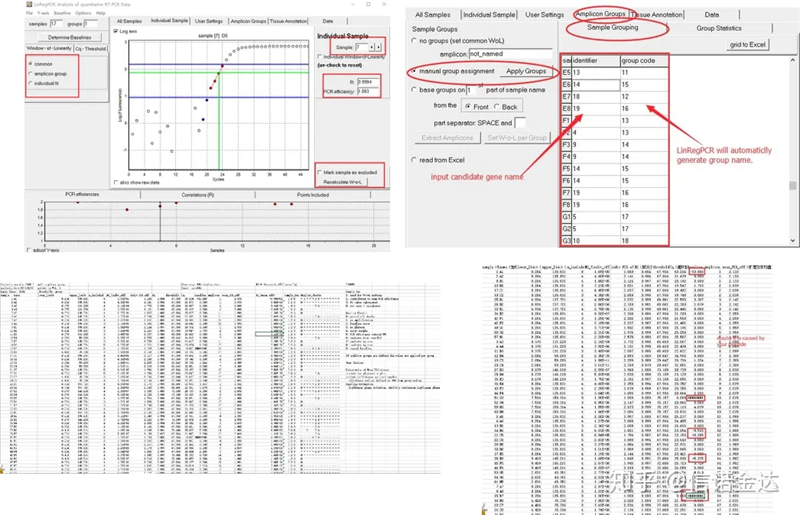
Fig. 8 Resultado y salida de datos
Proceso experimental:
Requisitos de calidad del ARN:
Pureza:1.72,0 indica que puede haber isotiocianato residual.El ácido nucleico limpio A260/A230 debe estar alrededor de 2. Si hay una fuerte absorción a 230 nm, indica que hay compuestos orgánicos como iones de fenato.Además, puede detectarse mediante electroforesis en gel de agarosa al 1,5%.Señale el marcador, porque el ssRNA no tiene desnaturalización y el logaritmo del peso molecular no tiene una relación lineal, y el peso molecular no se puede expresar correctamente.Concentración: Teóricamentenomenos de 100 ng/ul, si la concentración es demasiado baja, la pureza es generalmente baja, no alta
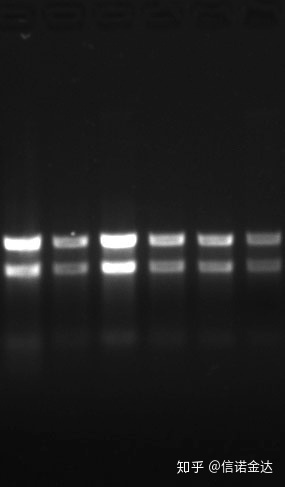
Gel de ARN Fig9
Además, si la muestra es valiosa y la concentración de ARN es alta, se recomienda dividirla en alícuotas después de la extracción y diluir el ARN a una concentración final de 100-300 ng/ul para la transcripción inversa.Enel proceso de transcripción inversa, cuando se transcribe el ARNm, se usan cebadores oligo (dt) que pueden unirse específicamente a las colas de poliA para la transcripción inversa, mientras que lncRNA y circRNA usan cebadores hexámeros aleatorios (Random 6 mer) para la transcripción inversa del ARN total. Para miARN, se usan cebadores de bucle de cuello específicos de miARN para la transcripción inversa.Muchas empresas ahora han lanzado kits especiales para relaves.Para el método de bucle de tallo, el método de seguimiento es más conveniente, de alto rendimiento y ahorra reactivos, pero el efecto de distinguir miARN de la misma familia no debería ser tan bueno como el método de bucle de tallo.Cada kit de transcripción inversa tiene requisitos para la concentración de cebadores específicos de genes (bucles de tallo).La referencia interna utilizada para miRNA es U6.En el proceso de inversión de bucle de vástago, un tubo de U6 debe invertirse por separado y los cebadores frontal y posterior de U6 deben agregarse directamente.Tanto circRNA como lncRNA pueden usar HKG como referencia interna.Endetección de ADNc,
si no hay problema con el ARN, el ADNc también debería estar bien.Sin embargo, si se persigue la perfección del experimento, lo mejor es utilizar un gen de referencia interno (gen de referencia, RG) que pueda distinguir gDNA de cds.En general, RG es un gen de limpieza., HKG) como se muestra en la Figura 10;En ese momento, estaba haciendo proteína de almacenamiento de soja y usaba intrones que contenían actina7 como referencia interna.El tamaño del fragmento amplificado de este cebador en gDNA fue de 452 pb, y si se usó cDNA como molde, fue de 142 pb.Luego, los resultados de la prueba encontraron que parte del cDNA en realidad estaba contaminado por gDNA, y también demostró que no había ningún problema con el resultado de la transcripción inversa, y que podría usarse como plantilla para PCR.Es inútil realizar electroforesis en gel de agarosa directamente con cDNA, y es una banda difusa, lo que no convence.
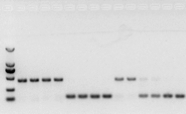
Figura 10 Detección de ADNc
La determinación de las condiciones de qPCRGeneralmente no hay problema de acuerdo con el protocolo del kit, principalmente en el paso de valor tm.Si algunos cebadores no están bien diseñados durante el diseño del cebador, lo que da como resultado una gran diferencia entre el valor de tm y los 60 °C teóricos, se recomienda que el cDNA Después de mezclar las muestras, ejecute una PCR en gradiente con los cebadores y trate de evitar establecer la temperatura sin bandas como el valor de TM.
Análisis de los datos
El método de procesamiento de PCR cuantitativa de fluorescencia relativa convencional es básicamente de acuerdo con 2-ΔΔCT.Plantilla de procesamiento de datos.
Productos relacionados:
PCR en tiempo real FácilTM –Taqman
PCR en tiempo real FácilTM –SYBR VERDE I
RT Easy I (Master Premix para la síntesis de la primera cadena de cDNA)
RT Easy II (Master Premix para la síntesis de ADNc de primera cadena para qPCR)
Hora de publicación: 14-mar-2023



