



La pérdida auditiva (HL) es la enfermedad de discapacidad sensorial más común en humanos.En los países desarrollados, alrededor del 80% de los casos de sordera prelingual en niños son causados por factores genéticos.Los más comunes son los defectos de un solo gen (como se muestra en la Fig. 1), se ha encontrado que 124 mutaciones genéticas están asociadas con la pérdida auditiva no sindrómica en humanos, el resto son causados por factores ambientales.Un implante coclear (un dispositivo electrónico colocado en el oído interno que proporciona estimulación eléctrica directamente al nervio auditivo) es, con diferencia, la opción más eficaz para tratar el HL grave, mientras que un audífono (un dispositivo electrónico externo que convierte y amplifica las ondas sonoras) puede ayudar a los pacientes con HL moderado.Sin embargo, actualmente no hay medicamentos disponibles para tratar el HL hereditario (GHL).En los últimos años, la terapia génica ha recibido una atención cada vez mayor como un enfoque prometedor para tratar la disfunción del oído interno.
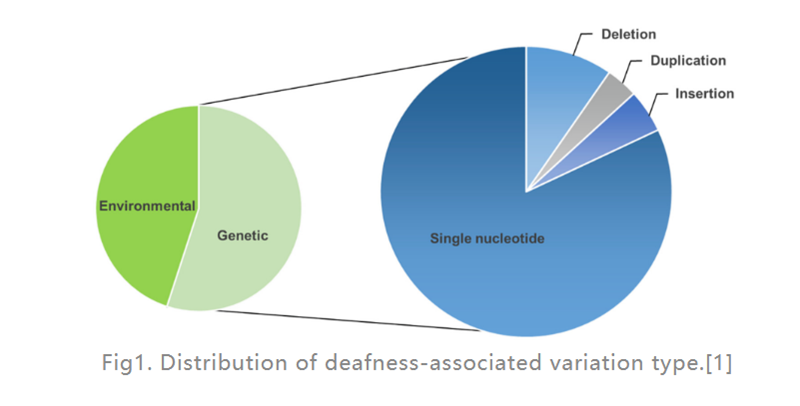
Figura 1.Distribución del tipo de variación asociado a la sordera.[1]
Recientemente, científicos del Instituto Salk y la Universidad de Sheffield publicaron un resultado de investigación en Molecular Therapy - Methods & Clinical Development [2], que mostró amplias perspectivas de aplicación para la terapia génica in vivo de la sordera hereditaria.Uri Manor, profesor asistente de investigación en el Instituto Salk y director del Centro Waitt de Biofotónica Avanzada, dijo que nació con una pérdida auditiva severa y sintió que restaurar la audición sería un regalo maravilloso.Su investigación anterior encontró que Eps8 es una proteína reguladora de actina con actividades de unión y protección de actina;en las células ciliadas cocleares, el complejo proteico formado por Eps8 con MYO15A, WHIRLIN, GPSM2 y GNAI3 existe principalmente en la mayoría de las puntas de los estereocilios largos, que junto con MYO15A localizan BAIAP2L2 en las puntas de los estereocilios más cortos, son necesarios para el mantenimiento de los haces de cabello.Por lo tanto, Eps8 puede regular la longitud de los estereocilios de las células ciliadas, lo cual es esencial para la función auditiva normal;La eliminación o mutación de Eps8 conducirá a estereocilios cortos, lo que hace que no pueda convertir correctamente el sonido en señales eléctricas para la percepción del cerebro, lo que a su vez conduce a la sordera..Al mismo tiempo, el colaborador Walter Marcotti, profesor de la Universidad de Sheffield, descubrió que las células ciliadas no pueden desarrollarse normalmente en ausencia de Eps8.En este estudio, Manor y Marcotti se unieron para investigar si agregar Eps8 a las células estereociliares podría restaurar su función y, a su vez, mejorar la audición en ratones.El equipo de investigación utilizó el vector del virus adenoasociado (AAV) Anc80L65 para administrar la secuencia de codificación que contenía EPS8 de tipo salvaje en la cóclea de ratones Eps8-/- recién nacidos P1-P2 mediante inyección de membrana de ventana redonda;en células ciliadas cocleares de ratón La función de los estereocilios se reparó antes de que maduraran;y el efecto de reparación se caracterizó mediante tecnología de imagen y medición de estereocilios.Los resultados mostraron que Eps8 aumentó la longitud de los estereocilios y restauró la función de las células ciliadas en las células de baja frecuencia.También encontraron que, con el tiempo, las células parecían perder su capacidad de ser rescatadas por esta terapia génica.La implicación es que es posible que este tratamiento deba administrarse en el útero, ya que las células ciliadas Eps8-/- pueden haber madurado o acumulado daños irreparables después del nacimiento de los ratones.“Eps8 es una proteína con muchas funciones diferentes y aún queda mucho por explorar”, dijo Manor.La investigación futura incluirá investigar el efecto de la terapia génica Eps8 en la restauración de la audición en diferentes etapas de desarrollo y si es posible prolongar las oportunidades de tratamiento.Coincidentemente, en noviembre de 2020, el profesor KarenB Avraham de la Universidad de Tel Aviv en Israel publicó sus resultados en la revista EMBO Molecular Medicine [3], utilizando una innovadora tecnología de terapia génica para crear un virus adenoasociado sintético inofensivo AAV9-PHP.B, El defecto genético en las células ciliadas de los ratones Syne4-/- se reparó mediante la inyección de un virus que portaba la secuencia de codificación de Syne4 en el oído interno de los ratones, lo que le permitió ingresar a las células ciliadas y liberar el material genético transportado, permitiéndoles madurar y funcionar normalmente (como en la Fig. 2).
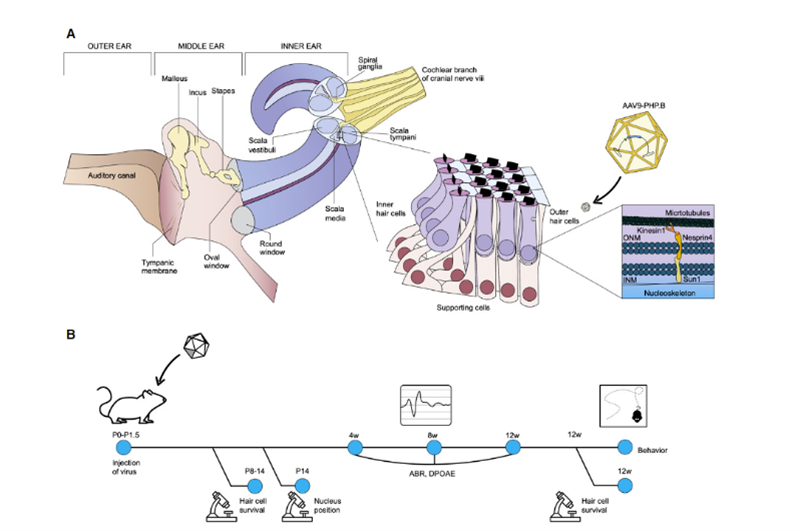
Figura 2.Representación esquemática de la anatomía del oído interno, con énfasis en el órgano de Corti y la función celular de nesprin-4.
Se puede ver que el uso de la terapia génica para lograr el propósito de tratar enfermedades hereditarias a nivel genético insertando, eliminando o corrigiendo cualquier gen mutado para el tratamiento (es decir, controlando los cambios genéticos en la enfermedad) tiene un alto efecto clínico.perspectivas de aplicación.Los métodos actuales de terapia génica para la sordera genéticamente deficiente se pueden dividir en las siguientes categorías:
reemplazo de genes
Podría decirse que el reemplazo de genes es la forma más "sencilla" de terapia génica, basada en identificar y reemplazar un gen defectuoso con una copia normal o de tipo salvaje del gen.Primer estudio exitoso de terapia génica del oído interno para la pérdida auditiva causada por la eliminación del gen del transportador vesicular de glutamato 3 (VGLUT3);La entrega mediada por AAV1 de la sobreexpresión exógena de VGLUT3 en las células ciliadas del oído interno (IHC) puede dar como resultado una recuperación auditiva sostenida, una recuperación parcial de la morfología sináptica de la cinta y respuestas convulsivas [4].Sin embargo, en los ejemplos que incluyen los dos reemplazos de genes proporcionados por AAV descritos en la introducción anterior, es importante tener en cuenta que los modelos de ratón utilizados para ciertos tipos de trastornos de pérdida de audición hereditaria por eliminación de genes son temporalmente diferentes de los humanos, y en los ratones P1, el oído interno está en la etapa madura de desarrollo.En contraste, los humanos nacen con un oído interno maduro.Esta diferencia impide la posible aplicación de los resultados del ratón al tratamiento de los trastornos de la sordera hereditaria humana, a menos que la terapia génica se administre a oídos de ratones maduros.
Edición de genes: CRISPR/Cas9
En comparación con el "reemplazo de genes", el desarrollo de la tecnología de edición de genes ha traído el amanecer del tratamiento de enfermedades genéticas desde la raíz.Es importante destacar que el método de edición de genes compensa las deficiencias de los métodos tradicionales de terapia génica de sobreexpresión que no son adecuados para las enfermedades de sordera hereditaria dominante, y el problema de que el método de sobreexpresión no dura mucho.Después de que los investigadores chinos eliminaran específicamente el alelo mutante Myo6C442Y en ratones Myo6WT/C442Y utilizando el sistema de edición de genes AAV-SaCas9-KKH-Myo6-g2, y dentro de los 5 meses posteriores a la eliminación, los ratones restauraron la función auditiva del modelo;al mismo tiempo, también se observó que mejoró la tasa de supervivencia de las células ciliadas en el oído interno, la forma de los cilios se volvió regular y se corrigieron los indicadores electrofisiológicos [5].Este es el primer estudio en el mundo que utiliza la tecnología CRISPR/Cas9 para el tratamiento de la sordera hereditaria causada por la mutación del gen Myo6, y es un avance importante en la investigación de la tecnología de edición de genes para el tratamiento de la sordera hereditaria.La traducción clínica del tratamiento proporciona una base científica sólida.
Métodos de administración de terapia génica
Para que la terapia génica tenga éxito, las moléculas de ADN desnudo no pueden penetrar en las células de manera efectiva debido a su hidrofilicidad y carga negativa de grupos fosfato, y para garantizar la integridad de las moléculas de ácido nucleico suplementadas, se debe seleccionar un método seguro y eficaz.El ADN suplementado se entrega a la célula o tejido objetivo.AAV se usa ampliamente como vehículo de administración para el tratamiento de enfermedades debido a su alto efecto infeccioso, baja inmunogenicidad y amplio tropismo a varios tipos de tejidos.En la actualidad, una gran cantidad de trabajo de investigación ha determinado el tropismo de diferentes subtipos de AAV en relación con diferentes tipos de células en la cóclea del ratón.El uso de las características de administración de AAV combinadas con promotores específicos de células puede lograr una expresión específica de células, lo que puede reducir los efectos fuera del objetivo.Además, como alternativa a los vectores AAV tradicionales, constantemente se desarrollan nuevos vectores AAV sintéticos que muestran una capacidad de transducción superior en el oído interno, de los cuales AAV2/Anc80L65 es el más utilizado.Los métodos de administración no viral se pueden dividir en métodos físicos (microinyección y electroporación) y métodos químicos (nanopartículas de oro, basadas en lípidos y basadas en polímeros).Ambos enfoques se han utilizado en el tratamiento de los trastornos de la sordera hereditaria y han mostrado diferentes ventajas y limitaciones.Además del vehículo de administración para la terapia génica como vehículo, se pueden emplear diferentes enfoques para la administración de genes in vivo en función de diferentes tipos de células diana, vías de administración y eficacia terapéutica.La intrincada estructura del oído interno dificulta el acceso a las células diana y la distribución de los agentes de edición del genoma es lenta.El laberinto membranoso se encuentra dentro del laberinto óseo del hueso temporal e incluye el conducto coclear, el conducto semicircular, el utrículo y el globo.Su aislamiento relativo, la circulación linfática mínima y la separación de la sangre por una barrera de laberinto de sangre limitan la administración sistémica efectiva de la terapia solo a los ratones recién nacidos.Para obtener títulos virales adecuados para la terapia génica, es necesaria la inyección local directa de vectores virales en el oído interno.Las rutas establecidas de inyección incluyen [6]: (1) membrana de ventana redonda (RWM), (2) traqueotomía, (3) cocleostomía endolinfática o perilinfática, (4) membrana de ventana redonda más fenestración de tubo (CF) (como en la Fig. 3).
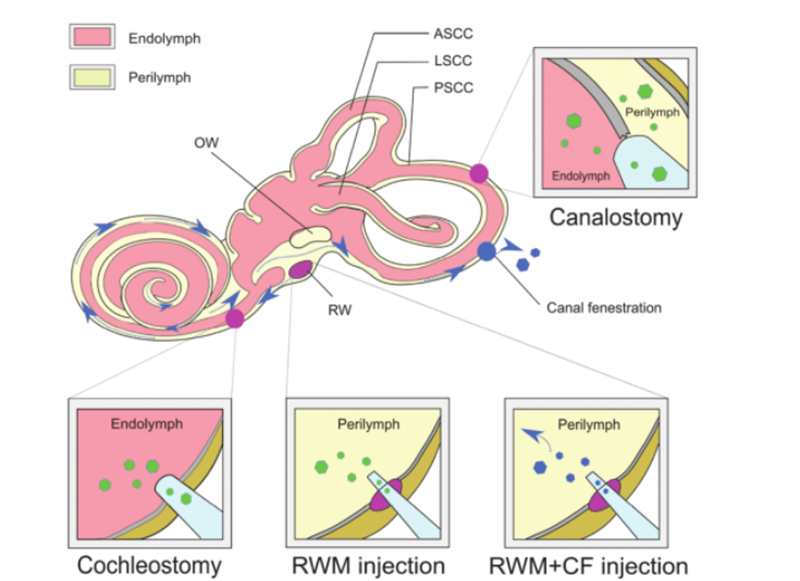
Fig. 3.Administración del oído interno de la terapia génica.
Aunque se han hecho muchos avances en la terapia génica, basados en objetivos de traslación clínica, se necesita más trabajo antes de que la terapia génica pueda convertirse en una opción de tratamiento de primera línea para pacientes con enfermedades genéticas, especialmente en el desarrollo de vectores y métodos de administración seguros y efectivos.Pero creemos que en un futuro cercano, este tipo de tratamientos se convertirán en un elemento básico de la terapia personalizada y tendrán un impacto muy positivo en la vida de las personas con trastornos genéticos y sus familias.
Foregene también ha lanzado un kit de detección de alto rendimiento para genes específicos, que es rápido y puede realizar reacciones de transcripción inversa y qPCR sin extracción de ARN.
Enlaces de productos
Kit Cell Direct RT-qPCR: Taqman/SYBR GREEN I
Para obtener más información sobre el producto, comuníquese con:
Hora de publicación: 02-sep-2022



