



- La PCR es un método utilizado para amplificar ADN a partir de una pequeña cantidad de plantilla de ADN.La RT-PCR utiliza la transcripción inversa para producir una plantilla de ADN a partir de una fuente de ARN que luego puede amplificarse.
- La PCR y la RT-PCR suelen ser reacciones de punto final, mientras que la qPCR y la RT-qPCR utilizan la cinética de la velocidad de síntesis del producto durante la reacción de PCR para cuantificar la cantidad de plantilla presente.
- Los métodos más nuevos, como la PCR digital, proporcionan una cuantificación absoluta de la plantilla de ADN inicial, mientras que métodos como la PCR isotérmica reducen la necesidad de equipos costosos para proporcionar resultados confiables.
La reacción en cadena de la polimerasa (PCR) es una técnica de biología molecular relativamente simple y ampliamente utilizada para amplificar y detectar secuencias de ADN y ARN.En comparación con los métodos tradicionales de clonación y amplificación de ADN, que a menudo pueden llevar días, la PCR requiere sólo unas pocas horas.La PCR es muy sensible y requiere una plantilla mínima para la detección y amplificación de secuencias específicas.Los métodos básicos de PCR han avanzado aún más desde la simple detección de ADN y ARN.A continuación, proporcionamos una descripción general de los diferentes métodos de PCR y los reactivos que proporcionamos en Enzo Life Sciences para sus necesidades de investigación.¡Nuestro objetivo es ayudar a los científicos a acceder rápidamente a reactivos de PCR para utilizarlos en su próximo proyecto de investigación!
PCR
Para la PCR estándar, todo lo que necesita es una ADN polimerasa, magnesio, nucleótidos, cebadores, la plantilla de ADN que se va a amplificar y un termociclador.El mecanismo de la PCR es tan simple como su propósito: 1) el ADN bicatenario (ADNbc) se desnaturaliza con calor, 2) los cebadores se alinean con las hebras individuales de ADN y 3) los cebadores se extienden mediante la ADN polimerasa, lo que da como resultado dos copias del ADN. cadena de ADN original.El proceso de desnaturalización, recocido y alargamiento a lo largo de una serie de temperaturas y tiempos se conoce como un ciclo de amplificación (Fig. 1).
|
|
| Figura 1.Representación esquemática de un ciclo de amplificación por PCR. |
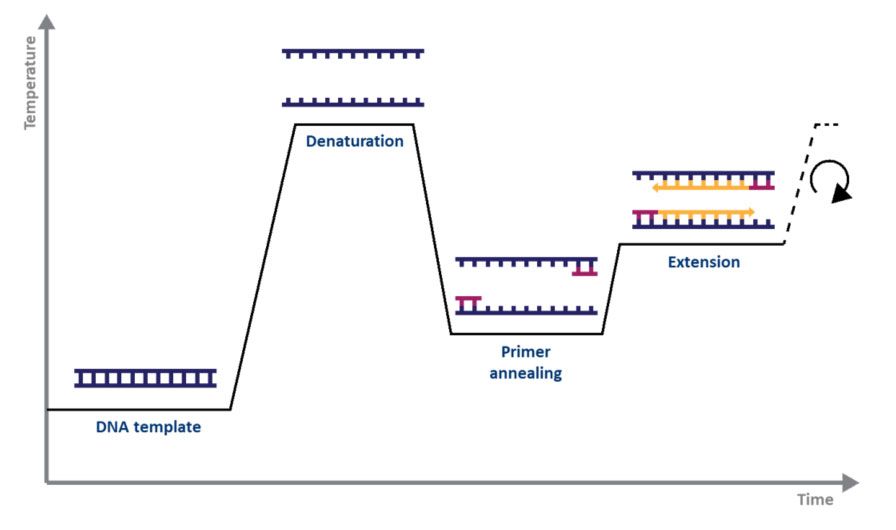
Cada paso del ciclo debe optimizarse para la plantilla y el conjunto de cebadores utilizados.Este ciclo se repite aproximadamente 20 a 40 veces y luego se puede analizar el producto amplificado, generalmente mediante gel de agarosa (Fig. 2).
| |
| Figura 2.Amplificación de un molde de ADN mediante PCR y análisis mediante electroforesis en gel de agarosa. |
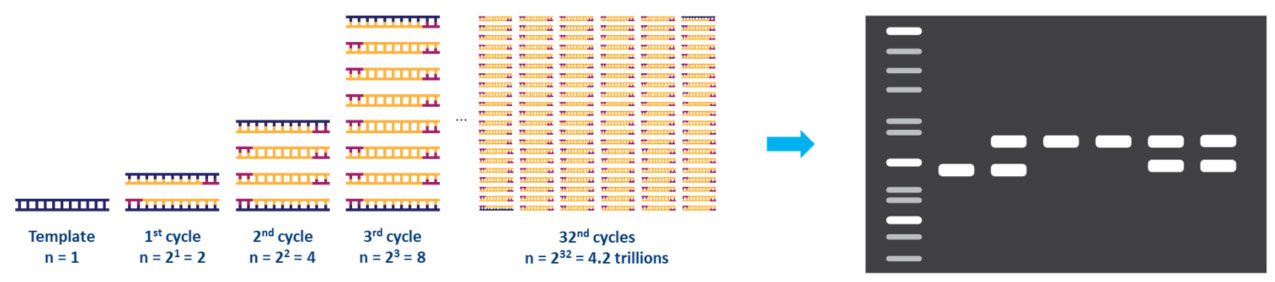
Como la PCR es un método muy sensible y se requieren volúmenes muy pequeños para reacciones individuales, se recomienda preparar una mezcla maestra para varias reacciones.La mezcla maestra debe mezclarse bien y luego dividirse por el número de reacciones, asegurando que cada reacción contendrá la misma cantidad de enzima, dNTP y cebadores.Muchos proveedores, como Enzo Life Sciences, también ofrecen mezclas de PCR que ya contienen todo excepto los cebadores y la plantilla de ADN.
Las regiones ricas en guanina/citosina (ricas en GC) representan un desafío en las técnicas de PCR estándar.Las secuencias ricas en GC son más estables que las secuencias con menor contenido de GC.Además, las secuencias ricas en GC tienden a formar estructuras secundarias, como bucles en horquilla.Como resultado, las dobles hebras ricas en GC son difíciles de separar completamente durante la fase de desnaturalización.En consecuencia, la ADN polimerasa no puede sintetizar la nueva cadena sin obstáculos.Una temperatura de desnaturalización más alta puede mejorar esto, y los ajustes hacia una temperatura de hibridación más alta y un tiempo de hibridación más corto pueden evitar la unión inespecífica de cebadores ricos en GC.Reactivos adicionales pueden mejorar la amplificación de secuencias ricas en GC.El DMSO, el glicerol y la betaína ayudan a alterar las estructuras secundarias causadas por las interacciones GC y, por lo tanto, facilitan la separación de las dobles hebras.
PCR de inicio en caliente
La amplificación inespecífica es un problema que puede ocurrir durante la PCR.La mayoría de las ADN polimerasas que se utilizan para la PCR funcionan mejor a temperaturas de entre 68 °C y 72 °C.Sin embargo, la enzima también puede ser activa a temperaturas más bajas, aunque en menor grado.A temperaturas muy por debajo de la temperatura de hibridación, los cebadores pueden unirse de forma no específica y provocar una amplificación no específica, incluso si la reacción se realiza en hielo.Esto se puede prevenir mediante el uso de inhibidores de la polimerasa que se disocian de la ADN polimerasa sólo una vez que se alcanza una determinada temperatura, de ahí el término PCR de inicio en caliente.El inhibidor puede ser un anticuerpo que se une a la polimerasa y se desnaturaliza a la temperatura de desnaturalización inicial (normalmente 95°C).
Polimerasa de alta fidelidad
Si bien las ADN polimerasas amplifican con bastante precisión la secuencia plantilla original, pueden ocurrir errores en la coincidencia de nucleótidos.Los desajustes en aplicaciones como la clonación pueden dar como resultado transcripciones truncadas y proteínas mal traducidas o inactivas en sentido posterior.Para evitar estos desajustes, se han identificado e incorporado al flujo de trabajo polimerasas con actividad de "corrección de pruebas".La primera polimerasa correctora, Pfu, fue identificada en 1991 en Pyrococcus furiosus.Esta enzima Pfu tiene una actividad exonucleasa de 3' a 5'.A medida que se amplifica el ADN, la exonucleasa elimina los nucleótidos no coincidentes en el extremo 3' de la cadena.Luego se reemplaza el nucleótido correcto y continúa la síntesis de ADN.La identificación de secuencias de nucleótidos incorrectas se basa en la afinidad de unión del nucleósido trifosfato correcto con la enzima, donde la unión ineficiente ralentiza la síntesis y permite el reemplazo correcto.La actividad correctora de la polimerasa Pfu produce menos errores en la secuencia final en comparación con la ADN polimerasa Taq.En los últimos años, se han identificado otras enzimas correctoras y se han realizado modificaciones de la enzima Pfu original para reducir aún más la tasa de error durante la amplificación del ADN.
RT-PCR
La PCR con transcripción inversa, o RT-PCR, permite el uso de ARN como plantilla.Un paso adicional permite la detección y amplificación de ARN.El ARN se transcribe de forma inversa en ADN complementario (ADNc) mediante la transcriptasa inversa.La calidad y pureza del molde de ARN son esenciales para el éxito de la RT-PCR.El primer paso de la RT-PCR es la síntesis de un híbrido de ADN/ARN.La transcriptasa inversa también tiene una función RNasa H, que degrada la porción de ARN del híbrido.La molécula de ADN monocatenaria se completa luego mediante la actividad ADN polimerasa dependiente de ADN de la transcriptasa inversa en ADNc.La eficiencia de la reacción de la primera cadena puede afectar el proceso de amplificación.A partir de ahora, se utiliza el procedimiento de PCR estándar para amplificar el ADNc.La posibilidad de revertir ARN en ADNc mediante RT-PCR tiene muchas ventajas y se utiliza principalmente para análisis de expresión génica.El ARN es monocatenario y muy inestable, lo que dificulta su trabajo.Suele servir como primer paso en qPCR, que cuantifica las transcripciones de ARN en una muestra biológica.
qPCR y RT-qPCR
La PCR cuantitativa (qPCR) se utiliza para detectar, caracterizar y cuantificar ácidos nucleicos para numerosas aplicaciones.En RT-qPCR, las transcripciones de ARN a menudo se cuantifican mediante transcripción inversa en ADNc primero, como se describió anteriormente, y luego se lleva a cabo la qPCR posteriormente.Como en la PCR estándar, el ADN se amplifica mediante tres pasos repetidos: desnaturalización, hibridación y elongación.Sin embargo, en qPCR, el etiquetado fluorescente permite la recopilación de datos a medida que avanza la PCR.Esta técnica tiene muchos beneficios debido a la variedad de métodos y químicas disponibles.
En la qPCR basada en colorante (normalmente verde), el marcaje fluorescente permite la cuantificación de las moléculas de ADN amplificadas mediante el uso de un colorante de unión a ADNds.Durante cada ciclo, se mide la fluorescencia.La señal de fluorescencia aumenta proporcionalmente a la cantidad de ADN replicado.Por tanto, el ADN se cuantifica en “tiempo real” (Fig. 3).Las desventajas de la qPCR basada en colorantes son que solo se puede examinar un objetivo a la vez y que el colorante se unirá a cualquier ADN bicatenario presente en la muestra.
|
|
| Figura 3.Amplificar una plantilla de ADN mediante qPCR y medir la señal de fluorescencia en tiempo real. |
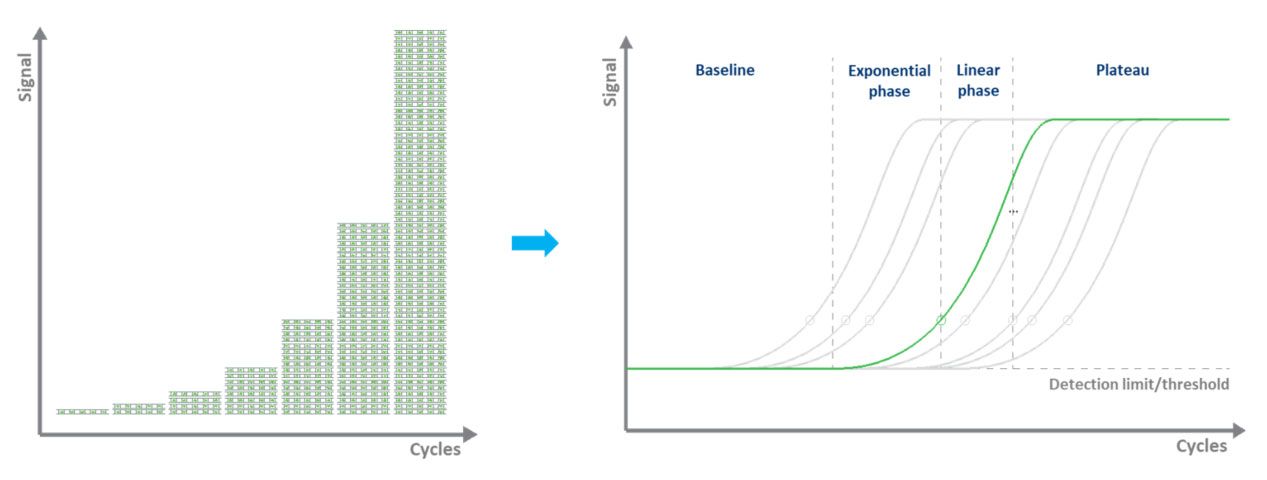
En la qPCR basada en sondas, se pueden detectar muchos objetivos simultáneamente en cada muestra, pero esto requiere la optimización y el diseño de una sonda específica del objetivo utilizada además de los cebadores.Hay varios tipos de diseños de sonda disponibles, pero el tipo más común es una sonda de hidrólisis, que incorpora un fluoróforo y un extintor.La transferencia de energía por resonancia de fluorescencia (FRET) evita la emisión del fluoróforo a través del extintor mientras la sonda está intacta.Sin embargo, durante la reacción de PCR, la sonda se hidroliza durante la extensión del cebador y la amplificación de la secuencia específica a la que está unida.La escisión de la sonda separa el fluoróforo del extintor y da como resultado un aumento de la fluorescencia dependiente de la amplificación (Fig. 4).Por lo tanto, la señal de fluorescencia de una reacción de qPCR basada en sonda es proporcional a la cantidad de secuencia objetivo de la sonda presente en la muestra.Debido a que la qPCR basada en sondas es más específica que la qPCR basada en colorantes, suele ser la tecnología utilizada en los ensayos de diagnóstico basados en qPCR.
| |
| Figura 4.Diferencias entre qPCR basada en colorantes y basada en sondas. |
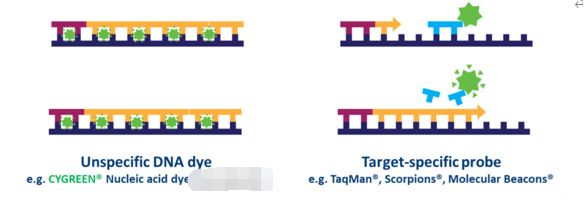
Amplificación isotérmica
Las técnicas de PCR mencionadas anteriormente requieren equipos de termociclado costosos para aumentar y disminuir con precisión las temperaturas de la cámara para los pasos de desnaturalización, recocido y extensión.Se han desarrollado varias técnicas que no necesitan dispositivos tan precisos y pueden realizarse en un simple baño de agua o incluso dentro de las células de interés.Estas técnicas se denominan colectivamente amplificación isotérmica y funcionan en base a amplificación exponencial, lineal o en cascada.
El tipo más conocido de amplificación isotérmica es la amplificación isotérmica mediada por bucle o LAMP.LAMP utiliza amplificación exponencial a 65⁰C para amplificar ADN o ARN molde.Al realizar LAMP, se utilizan de cuatro a seis cebadores complementarios a regiones del ADN objetivo con una ADN polimerasa para sintetizar ADN nuevo.Dos de estos cebadores tienen secuencias complementarias que reconocen secuencias en los otros cebadores y se unen a ellas, lo que permite que se forme una estructura de "bucle" en el ADN recién sintetizado que luego ayuda a la hibridación de los cebadores en rondas posteriores de amplificación.LAMP se puede visualizar mediante múltiples métodos, incluida la fluorescencia, la electroforesis en gel de agarosa o la colorimetría.La facilidad para visualizar y detectar la presencia o ausencia del producto mediante colorimetría y la falta de equipos costosos necesarios hicieron de LAMP una opción adecuada para las pruebas de SARS-CoV-2 en áreas donde las pruebas de laboratorio clínico no estaban disponibles, o el almacenamiento y transporte de muestras. no era factible, o en laboratorios que anteriormente no contaban con equipos de termociclado de PCR.
Hora de publicación: 19-ago-2023



