



La tecnología de diagnóstico molecular utiliza métodos de biología molecular para detectar la expresión y estructura del material genético del cuerpo humano y varios patógenos, a fin de lograr el propósito de predecir y diagnosticar enfermedades.
En los últimos años, con la actualización y la iteración de la tecnología de diagnóstico molecular, la aplicación clínica del diagnóstico molecular se ha vuelto cada vez más extensa y profunda, y el mercado de diagnóstico molecular ha entrado en un período de rápido desarrollo.
El autor resume las tecnologías de diagnóstico molecular comunes en el mercado y se divide en tres partes: la primera parte presenta la tecnología PCR, la segunda parte presenta la tecnología de amplificación isotérmica de ácidos nucleicos y la segunda parte presenta la tecnología de secuenciación.
01
Parte I: Tecnología PCR
tecnología PCR
La PCR (reacción en cadena de la polimerasa) es una de las tecnologías de amplificación de ADN in vitro, con una historia de más de 30 años.
La tecnología PCR fue iniciada en 1983 por Kary Mullis de Cetus, EE. UU.Mullis solicitó una patente de PCR en 1985 y publicó el primer artículo académico de PCR sobre ciencia ese mismo año.Mullis ganó el Premio Nobel de Química en 1993.
Principios básicos de PCR
La PCR puede amplificar fragmentos de ADN diana más de un millón de veces.El principio es que, bajo la catálisis de la ADN polimerasa, la hebra original de ADN se utiliza como molde y un cebador específico se utiliza como punto de partida para la extensión.Se replica in vitro a través de pasos como la desnaturalización, el recocido y la extensión.El proceso del ADN de la hebra hija complementario al ADN molde de la hebra progenitora.

El proceso de PCR estándar se divide en tres pasos:
1. Desnaturalización: use altas temperaturas para separar las dobles cadenas de ADN.Los enlaces de hidrógeno entre las cadenas dobles de ADN se rompen a altas temperaturas (93-98°C).
2. Recocido: después de separar el ADN de doble cadena, se baja la temperatura para que el cebador pueda unirse al ADN de cadena sencilla.
3. Extensión: la ADN polimerasa comienza a sintetizar cadenas complementarias a lo largo de las cadenas de ADN a partir de los cebadores unidos cuando se baja la temperatura.Cuando se completa la extensión, se completa un ciclo y el número de fragmentos de ADN se duplica.
Alternando estos tres pasos 25-35 veces, el número de fragmentos de ADN aumentará exponencialmente.

El ingenio de la PCR es que se pueden diseñar diferentes cebadores para diferentes genes objetivo, de modo que los fragmentos del gen objetivo se puedan amplificar en un corto período de tiempo.
Hasta ahora, la PCR se puede dividir en tres categorías, a saber, la PCR ordinaria, la PCR cuantitativa fluorescente y la PCR digital.
La primera generación de PCR ordinaria
Use un instrumento de amplificación de PCR ordinario para amplificar el gen objetivo y luego use electroforesis en gel de agarosa para detectar el producto, solo se puede realizar un análisis cualitativo.
Las principales desventajas de la PCR de primera generación:
- Propenso a la amplificación no específica y resultados falsos positivos.
-La detección lleva mucho tiempo y la operación es engorrosa.
-Solo se pueden hacer pruebas cualitativas.
PCR cuantitativa de fluorescencia de segunda generación
La PCR cuantitativa de fluorescencia (PCR en tiempo real), también conocida como qPCR, se utiliza para monitorear la acumulación de productos amplificados a través de la acumulación de señales fluorescentes agregando sondas fluorescentes que pueden indicar el progreso del sistema de reacción y para juzgar los resultados a través de la curva de fluorescencia, y se puede cuantificar con la ayuda del valor Cq y la curva estándar.
Debido a que la tecnología qPCR se lleva a cabo en un sistema cerrado, la probabilidad de contaminación se reduce y la señal de fluorescencia se puede monitorear para la detección cuantitativa, por lo que es la más utilizada en la práctica clínica y se ha convertido en la tecnología dominante en PCR.
Las sustancias fluorescentes utilizadas en la PCR cuantitativa fluorescente en tiempo real se pueden dividir en: sondas fluorescentes TaqMan, balizas moleculares y colorantes fluorescentes.
1) Sonda fluorescente TaqMan:
Durante la amplificación por PCR, se agrega una sonda fluorescente específica mientras se agrega un par de cebadores.La sonda es un oligonucleótido y los dos extremos están marcados respectivamente con un grupo fluorescente informador y un grupo fluorescente extintor.
Cuando la sonda está intacta, la señal fluorescente emitida por el grupo informador es absorbida por el grupo desactivador;Durante la amplificación por PCR, la actividad de exonucleasa 5′-3′ de la enzima Taq escinde y degrada la sonda, lo que hace que el grupo fluorescente indicador y el extintor se separen, de modo que el sistema de monitoreo de fluorescencia pueda recibir la señal de fluorescencia, es decir, cada vez que se amplifica una cadena de ADN, se forma una molécula fluorescente y la acumulación de la señal de fluorescencia está completamente sincronizada con la formación del producto de PCR.
2) Tintes fluorescentes SYBR:
En el sistema de reacción de PCR, se agrega un exceso de colorante fluorescente SYBR.Después de que el colorante fluorescente SYBR se incorpora de forma no específica a la cadena doble de ADN, emite una señal fluorescente.La molécula de colorante SYBR que no se incorpora a la cadena no emitirá ninguna señal fluorescente, lo que garantiza que la señal fluorescente esté completamente sincronizada con el aumento de productos de PCR.SYBR solo se une al ADN de doble cadena, por lo que la curva de fusión se puede utilizar para determinar si la reacción de PCR es específica.


3) balizas moleculares
Es una sonda de oligonucleótidos de doble marcaje de tallo y bucle que forma una estructura de horquilla de aproximadamente 8 bases en los extremos 5 y 3.Las secuencias de ácido nucleico en ambos extremos se emparejan de manera complementaria, lo que hace que el grupo fluorescente y el grupo de extinción estén unidos.Cierra, no producirá fluorescencia.
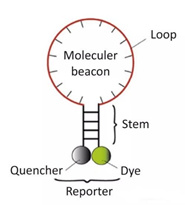
Después de que se genera el producto de PCR, durante el proceso de hibridación, la parte media de la baliza molecular se empareja con una secuencia de ADN específica y el gen fluorescente se separa del gen extintor para producir fluorescencia.

Las principales desventajas de la PCR de segunda generación:
Todavía falta sensibilidad y la detección de muestras con pocas copias no es precisa.
Hay una influencia del valor de fondo y el resultado es susceptible a interferencias.
PCR digital de tercera generación
La PCR digital (DigitalPCR, dPCR, Dig-PCR) calcula el número de copias de la secuencia objetivo a través de la detección del punto final y puede realizar una detección cuantitativa absoluta precisa sin usar controles internos ni curvas estándar.
La PCR digital utiliza detección de punto final y no depende del valor Ct (umbral del ciclo), por lo que la reacción de PCR digital se ve menos afectada por la eficiencia de amplificación y se mejora la tolerancia a los inhibidores de la reacción de PCR, con alta precisión y reproducibilidad.
Debido a las características de alta sensibilidad y alta precisión, los inhibidores de la reacción de PCR no interfieren fácilmente, y puede lograr una cuantificación absoluta verdadera sin productos estándar, lo que se ha convertido en un punto crítico de investigación y aplicación.
Según las diferentes formas de la unidad de reacción, se puede dividir en tres tipos: sistemas de microfluidos, chips y gotitas.
Hora de publicación: 08-jul-2021



